 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Carola Förster | + 2943 word(s) | 2943 | 2021-06-21 05:54:03 | | | |
| 2 | Bruce Ren | -21 word(s) | 2922 | 2021-06-21 09:59:27 | | |
Video Upload Options
Alzheimer’s disease (AD), the most common cause of dementia in the elderly, is a neurodegenerative disorder associated with neurovascular dysfunction and cognitive decline. While the deposition of amyloid β peptide (Aβ) and the formation of neurofibrillary tangles (NFTs) are the pathological hallmarks of AD-affected brains, the majority of cases exhibits a combination of comorbidities that ultimately lead to multi-organ failure. Of particular interest, it can be demonstrated that Aβ pathology is present in the hearts of patients with AD, while the formation of NFT in the auditory system can be detected much earlier than the onset of symptoms. Progressive hearing impairment may beget social isolation and accelerate cognitive decline and increase the risk of developing dementia.
1. Introduction: Alzheimer’s—A Systemic Disease?
2. The Blood–Brain Barrier
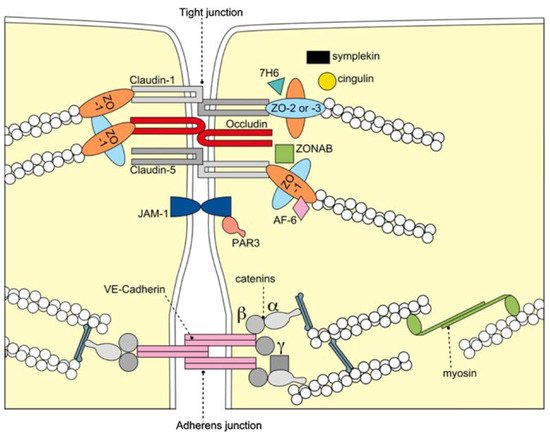
BBB Dysfunction and Vascular Risk Factors for AD
3. Amyloid Cardiomyopathy
References
- Sagare, A.P.; Bell, R.D.; Zlokovic, B.V. Neurovascular dysfunction and faulty amyloid beta-peptide clearance in Alzheimer disease. Cold Spring Harb. Perspect. Med. 2012, 2, a011452.
- Swanberg, M.M.; Tractenberg, R.E.; Mohs, R.; Thal, L.J.; Cummings, J.L. Executive Dysfunction in Alzheimer Disease. Arch. Neurol. 2004, 61, 556–560.
- Querfurth, H.W.; LaFerla, F.M. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344.
- Ballatore, C.; Lee, V.M.; Trojanowski, J.Q. Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders. Nat. Rev. Neurosci. 2007, 8, 663–672.
- Zhao, L.H. Alzheimer’s disease facts and figures. Alzheimer’s Dement. 2020, 16, 391–460.
- Du, X.; Wang, X.; Geng, M. Alzheimer’s disease hypothesis and related therapies. Transl. Neurodegener. 2018, 7, 1–7.
- Cummings, J.L. Alzheimer’s disease. N. Engl. J. Med. 2004, 351, 56–67.
- Hardy, J.; Selkoe, D.J. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science 2002, 297, 353–356.
- Bell, R.D.; Zlokovic, B.V. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009, 118, 103–113.
- Deane, R.; Yan, S.D.; Submamaryan, R.K.; LaRue, B.; Jovanovic, S.; Hogg, E.; Welch, D.; Manness, L.; Lin, C.; Yu, J.; et al. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med. 2003, 9, 907–913.
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-beta and neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 20021–20026.
- Meyer-Luehmann, M.; Coomaraswamy, J.; Bolmont, T.; Kaeser, S.; Schaefer, C.; Kilger, E.; Neuenschwander, A.; Abramowski, D.; Frey, P.; Jaton, A.L.; et al. Exogenous induction of cerebral beta-amyloidogenesis is governed by agent and host. Science 2006, 313, 1781–1784.
- Meyer-Luehmann, M.; Spires-Jones, T.L.; Prada, C.; Garcia-Alloza, M.; de Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-beta plaques in a mouse model of Alzheimer’s disease. Nature 2008, 451, 720–724.
- Eisele, Y.S.; Obermuller, U.; Heilbronner, G.; Baumann, F.; Kaeser, S.A.; Wolburg, H.; Walker, L.C.; Staufenbiel, M.; Heikenwalder, M.; Jucker, M. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science 2010, 330, 980–982.
- Prusiner, S.B. Human prion diseases and neurodegeneration. Curr. Top. Microbiol. Immunol. 1996, 207, 1–17.
- Zlokovic, B.V. New therapeutic targets in the neurovascular pathway in Alzheimer’s disease. Neurother. J. Am. Soc. Exp. Neurother. 2008, 5, 409–414.
- Zlokovic, B.V. Neurovascular pathways to neurodegeneration in Alzheimer’s disease and other disorders. Nat. Rev. Neurosci. 2011, 12, 723–738.
- De la Torre, J.C. The vascular hypothesis of Alzheimer’s disease: Bench to bedside and beyond. Neuro-Degener. Dis. 2010, 7, 116–121.
- Marchesi, V.T. Alzheimer’s dementia begins as a disease of small blood vessels, damaged by oxidative-induced inflammation and dysregulated amyloid metabolism: Implications for early detection and therapy. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2011, 25, 5–13.
- Zlokovic, B.V. Neurovascular mechanisms of Alzheimer’s neurodegeneration. Trends Neurosci. 2005, 28, 202–208.
- Przybyłowska, M.; Dzierzbicka, K.; Kowalski, S.; Chmielewska, K.; Inkielewicz-Stepniak, I. Therapeutic Potential of Multifunctional Derivatives of Cholinesterase Inhibitors. Curr. Neuropharmacol. 2020, 19, 1–24.
- Francis, P.T.; Palmer, A.M.; Snape, M.; Wilcock, G.K. The cholinergic hypothesis of Alzheimer’s disease: A review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 66, 137–147.
- Leng, F.; Edison, P. Neuroinflammation and microglial activation in Alzheimer disease: Where do we go from here? Nat. Rev. Neurol. 2020, 17, 157–172.
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Neurosci. 2018, 12, 25.
- Banks, W.A. From blood-brain barrier to blood-brain interface: New opportunities for CNS drug delivery. Nat. Rev. Drug Discov. 2016, 15, 275–292.
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596.
- Abbott, N.J. Blood-brain barrier structure and function and the challenges for CNS drug delivery. J. Inherit. Metab. Dis. 2013, 36, 437–449.
- Shityakov, S.; Salvador, E.; Pastorin, G.; Forster, C. Blood-brain barrier transport studies, aggregation, and molecular dynamics simulation of multiwalled carbon nanotube functionalized with fluorescein isothiocyanate. Int. J. Nanomed. 2015, 10, 1703–1713.
- Helms, H.C.; Abbott, N.J.; Burek, M.; Cecchelli, R.; Couraud, P.-O.; Deli, M.A.; Förster, C.; Galla, H.J.; Romero, I.; Shusta, E.V.; et al. In vitro models of the blood–brain barrier: An overview of commonly used brain endothelial cell culture models and guidelines for their use. Br. J. Pharmacol. 2016, 36, 862–890.
- McConnell, H.; Li, Z.; Woltjer, R.L.; Mishra, A. Astrocyte dysfunction and neurovascular impairment in neurological disorders: Correlation or causation? Neurochem. Int. 2019, 128, 70–84.
- Greenberg, S.M.; Gurol, E.; Rosand, J.; Smith, E.E. Amyloid Angiopathy-Related Vascular Cognitive Impairment. Stroke 2004, 35, 2616–2619.
- DeSimone, C.V.; Graff-Radford, J.; El-Harasis, M.A.; Rabinstein, A.A.; Asirvatham, S.J.; Holmes, D.R. Cerebral amyloid angiopathy and implications for atrial fibrillation management. Lancet 2017, 390, 9–11.
- Forster, C. Tight junctions and the modulation of barrier function in disease. Histochem. Cell Biol. 2008, 130, 55–70.
- Kinney, J.W.; BeMiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer’s Dementia: Transl. Res. Clin. Interv. 2018, 4, 575–590.
- Engelhardt, B.; Ransohoff, R.M. Capture, crawl, cross: The T cell code to breach the blood-brain barriers. Trends Immunol. 2012, 33, 579–589.
- Salvador, E.; Burek, M.; Forster, C.Y. Tight Junctions and the Tumor Microenvironment. Curr. Pathobiol. Rep. 2016, 4, 135–145.
- Kleinschnitz, C.; Blecharz, K.; Kahles, T.; Schwarz, T.; Kraft, P.; Göbel, K.; Meuth, S.G.; Burek, M.; Thum, T.; Stoll, G.; et al. Glucocorticoid Insensitivity at the Hypoxic Blood–Brain Barrier Can Be Reversed by Inhibition of the Proteasome. Stroke 2011, 42, 1081–1089.
- Yamazaki, Y.; Kanekiyo, T. Blood-Brain Barrier Dysfunction and the Pathogenesis of Alzheimer’s Disease. Int. J. Mol. Sci. 2017, 18, 1965.
- Hartz, A.M.; Bauer, B.; Soldner, E.L.; Wolf, A.; Boy, S.; Backhaus, R.; Mihaljevic, I.; Bogdahn, U.; Klunemann, H.H.; Schuierer, G.; et al. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke 2012, 43, 514–523.
- Atwood, C.S.; Martins, R.N.; Smith, M.A.; Perry, G. Senile plaque composition and posttranslational modification of amyloid-beta peptide and associated proteins. Peptides 2002, 23, 1343–1350.
- Kumar-Singh, S.; Pirici, D.; McGowan, E.; Serneels, S.; Ceuterick, C.; Hardy, J.; Duff, K.; Dickson, D.; Van Broeckhoven, C. Dense-Core Plaques in Tg2576 and PSAPP Mouse Models of Alzheimer’s Disease Are Centered on Vessel Walls. Am. J. Pathol. 2005, 167, 527–543.
- Hellberg, S.; Silvola, J.M.; Liljenbäck, H.; Kiugel, M.; Eskola, O.; Hakovirta, H.; Hörkkö, S.; Morisson-Iveson, V.; Hirani, E.; Saukko, P.; et al. Amyloid-Targeting PET Tracer [18F]Flutemetamol Accumulates in Atherosclerotic Plaques. Molecules 2019, 24, 1072.
- Van Assema, D.M.; Lubberink, M.; Rizzu, P.; Van Swieten, J.C.; Schuit, R.C.; Eriksson, J.; Scheltens, P.; Koepp, M.; Lammertsma, A.A.; Van Berckel, B.N. Blood–brain barrier P-glycoprotein function in healthy subjects and Alzheimer’s disease patients: Effect of polymorphisms in the ABCB1 gene. EJNMMI Res. 2012, 2, 57.
- Wolf, A.; Bauer, B.; Hartz, A.M. ABC Transporters and the Alzheimer’s Disease Enigma. Front. Psychiatry 2012, 3, 54.
- Carrano, A.; Hoozemans, J.J.; van der Vies, S.M.; Rozemuller, A.J.; van Horssen, J.; de Vries, H.E. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid. Redox Signal. 2011, 15, 1167–1178.
- Grammas, P.; Samany, P.G.; Thirumangalakudi, L. Thrombin and inflammatory proteins are elevated in Alzheimer’s disease microvessels: Implications for disease pathogenesis. J. Alzheimer’s Dis. 2006, 9, 51–58.
- Goos, J.D.; Kester, M.I.; Barkhof, F.; Klein, M.; Blankenstein, M.A.; Scheltens, P.; van der Flier, W.M. Patients with Alzheimer disease with multiple microbleeds: Relation with cerebrospinal fluid biomarkers and cognition. Stroke 2009, 40, 3455–3460.
- Lanfranconi, S.; Franco, G.; Borellini, L.; Denaro, F.; Basilico, P.; Parati, E.; Micieli, G.; Bersano, A. Genetics of cerebral hemorrhage and microbleeds. Panminerva Med. 2013, 55, 11–28.
- Brun, A.; Englund, E. A white matter disorder in dementia of the Alzheimer type: A pathoanatomical study. Ann. Neurol. 1986, 19, 253–262.
- Deane, R.; Wu, Z.; Sagare, A.; Davis, J.; Yan, S.D.; Hamm, K.; Xu, F.; Parisi, M.; LaRue, B.; Hu, H.W.; et al. LRP/amyloid beta-peptide interaction mediates differential brain efflux of Abeta isoforms. Neuron 2004, 43, 333–344.
- Bell, L.N.; Lee, L.; Saxena, R.; Bemis, K.G.; Wang, M.; Theodorakis, J.L.; Vuppalanchi, R.; Alloosh, M.; Sturek, M.; Chalasani, N. Serum proteomic analysis of diet-induced steatohepatitis and metabolic syndrome in the Ossabaw miniature swine. Am. J. Physiol. Liver Physiol. 2010, 298, G746–G754.
- Jellinger, K.A. Prevalence and impact of cerebrovascular lesions in Alzheimer and lewy body diseases. Neuro-Degener. Dis. 2010, 7, 112–115.
- Ruitenberg, A.; Heijer, T.D.; Bakker, S.L.M.; Van Swieten, J.C.; Koudstaal, P.J.; Hofman, A.; Breteler, M.M. Cerebral hypoperfusion and clinical onset of dementia: The Rotterdam study. Ann. Neurol. 2005, 57, 789–794.
- Vermeer, S.E.; Prins, N.D.; Heijer, T.D.; Hofman, A.; Koudstaal, P.J.; Breteler, M.M. Silent Brain Infarcts and the Risk of Dementia and Cognitive Decline. N. Engl. J. Med. 2003, 348, 1215–1222.
- Snowdon, D.A.; Greiner, L.H.; Mortimer, J.A.; Riley, K.P.; Greiner, P.A.; Markesbery, W.R. Brain Infarction and the Clinical Expression of Alzheimer DiseaseThe Nun Study. JAMA 1997, 277, 813–817.
- Jellinger, K. Cerebellar involvement in progressive supranuclear palsy. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1104–1105.
- Wendell, C.R.; Waldstein, S.R.; Ferrucci, L.; O’Brien, R.J.; Strait, J.B.; Zonderman, A.B. Carotid atherosclerosis and prospective risk of dementia. Stroke 2012, 43, 3319–3324.
- Di Marco, L.Y.; Venneri, A.; Farkas, E.; Evans, P.C.; Marzo, A.; Frangi, A. Vascular dysfunction in the pathogenesis of Alzheimer’s disease—A review of endothelium-mediated mechanisms and ensuing vicious circles. Neurobiol. Dis. 2015, 82, 593–606.
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56.
- Deane, R.; Bell, R.D.; Sagare, A.; Zlokovic, B.V. Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 2009, 8, 16–30.
- Troncone, L.; Luciani, M.; Coggins, M.; Wilker, E.H.; Ho, C.Y.; Codispoti, K.E.; Frosch, M.P.; Kayed, R.; del Monte, F. Abeta Amyloid Pathology Affects the Hearts of Patients With Alzheimer’s Disease: Mind the Heart. J. Am. Coll. Cardiol. 2016, 68, 2395–2407.
- Petrovitch, H.; White, L.; Izmirilian, G.; Ross, G.; Havlik, R.; Markesbery, W.; Nelson, J.; Davis, D.; Hardman, J.; Foley, D.; et al. Midlife blood pressure and neuritic plaques, neurofibrillary tangles, and brain weight at death: The HAAS☆. Neurobiol. Aging 2000, 21, 57–62.
- Tzourio, C. Hypertension, cognitive decline, and dementia: An epidemiological perspective. Dialogues Clin. Neurosci. 2007, 9, 61–70.
- Chakraborty, A.; de Wit, N.M.; van der Flier, W.M.; de Vries, H.E. The blood brain barrier in Alzheimer’s disease. Vasc. Pharmacol. 2017, 89, 12–18.
- Touyz, R.M. Oxidative stress and vascular damage in hypertension. Curr. Hypertens. Rep. 2000, 2, 98–105.
- Roberts, R.O.; Geda, Y.E.; Knopman, D.S.; Cha, R.H.; Boeve, B.F.; Ivnik, R.J.; Pankratz, V.S.; Tangalos, E.G.; Petersen, R.C. Metabolic syndrome, inflammation, and nonamnestic mild cognitive impairment in older persons: A population-based study. Alzheimer Dis. Assoc. Disord. 2010, 24, 11–18.
- Dar, T.; Sheikh, I.; Ganie, S.; Ali, R.; Singh, L.; Gan, S.H.; Kamal, M.A.; Zargar, M. Molecular Linkages Between Diabetes and Alzheimer’s Disease: Current Scenario and Future Prospects. CNS Neurol. Disord. Drug Targets 2014, 13, 290–298.
- Ott, A.; Stolk, R.; Van Harskamp, F.; Pols, H.A.P.; Hofman, A.; Breteler, M.M. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999, 53, 1937.
- Muhammad, S.; Bierhaus, A.; Schwaninger, M. Reactive oxygen species in diabetes-induced vascular damage, stroke, and Alzheimer’s disease. J. Alzheimer’s Dis. JAD 2009, 16, 775–785.
- Whitmer, R.A.; Gustafson, D.R.; Barrett-Connor, E.; Haan, M.N.; Gunderson, E.P.; Yaffe, K. Central obesity and increased risk of dementia more than three decades later. Neurology 2008, 71, 1057–1064.
- Grundy, S.M. Metabolic syndrome update. Trends Cardiovasc. Med. 2016, 26, 364–373.
- Nguyen, J.C.; Killcross, A.S.; Jenkins, T.A. Obesity and cognitive decline: Role of inflammation and vascular changes. Front. Neurosci. 2014, 8, 375.
- Swerdlow, R.H.; Khan, S.M. A “mitochondrial cascade hypothesis” for sporadic Alzheimer’s disease. Med. Hypotheses 2004, 63, 8–20.
- Wood, W.G.; Eckert, G.P.; Igbavboa, U.; Müller, W.E. Amyloid beta-protein interactions with membranes and cholesterol: Causes or casualties of Alzheimer’s disease. Biochim. Biophys. Acta (BBA) Biomembr. 2003, 1610, 281–290.
- Dempsey, R.J.; Vemuganti, R.; Varghese, T.; Hermann, B.P. A review of carotid atherosclerosis and vascular cognitive decline: A new understanding of the keys to symptomology. Neurosurgery 2010, 67, 484–493.
- Masoudi, F.A.; Rumsfeld, J.S.; Havranek, E.P.; House, J.A.; Peterson, E.D.; Krumholz, H.M.; Spertus, J.A. Age, functional capacity, and health-related quality of life in patients with heart failure. J. Card. Fail. 2004, 10, 368–373.
- Rich, M.W.; Goyal, P.; Forman, D.E. Age and Heart Failure Trials—Lessons from DAPA-HF. J. Card. Fail. 2020, 26, 191–192.
- Ihne, S.; Morbach, C.; Obici, L.; Palladini, G.; Störk, S. Amyloidosis in Heart Failure. Curr. Hear. Fail. Rep. 2019, 16, 285–303.
- Debette, S.; Bauters, C.; Leys, D.; Lamblin, N.; Pasquier, F.; de Groote, P. Prevalence and Determinants of Cognitive Impairment in Chronic Heart Failure Patients. Congest. Hear. Fail. 2007, 13, 205–208.
- Moreira, P.I.; Smith, M.A.; Zhu, X.; Nunomura, A.; Castellani, R.J.; Perry, G. Oxidative Stress and Neurodegeneration. Ann. N. Y. Acad. Sci. 2005, 1043, 545–552.
- Shin, S.C.; Robinson-Papp, J. Amyloid Neuropathies. Mt. Sinai J. Med. A J. Transl. Pers. Med. 2012, 79, 733–748, .
- Liao, R.; Ward, J. Amyloid Cardiomyopathy. Circ. Res. 2017, 120, 1865–1867, .
- Jensen-Dahm, C.; Waldemar, G.; Jensen, T.S.; Malmqvist, L.; Moeller, M.M.; Andersen, B.B.; Høgh, P.; Ballegaard, M. Autonomic Dysfunction in Patients with Mild to Moderate Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 681–689, .
- Serhiyenko, V.A. Cardiac autonomic neuropathy: Risk factors, diagnosis and treatment. World J. Diabetes 2018, 9, 1–24, .
- Sainz, A.L.; Moral, F.J.D.H.-D.; Dominguez, F.; Restrepo-Cordoba, A.; Amor-Salamanca, A.; Hernandez-Hernandez, A.; Ruiz-Guerrero, L.; Krsnik, I.; Cobo-Marcos, M.; Castro, V.; et al. Prevalence of cardiac amyloidosis among elderly patients with systolic heart failure or conduction disorders. Amyloid 2019, 26, 156–163, .
- Toledo, J.B.; Toledo, E.; Weiner, M.W.; Jack, C.R.; Jagust, W., Jr.; Lee, V.M.; Shaw, L.M.; Trojanowski, J.Q. Cardiovascular risk factors, cortisol, and amyloid-beta deposition in Alzheimer’s Disease Neuroimaging Initiative. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2012, 8, 483–489.
- Sanna, G.D.; Nusdeo, G.; Piras, M.R.; Forteleoni, A.; Murru, M.R.; Saba, P.S.; Dore, S.; Sotgiu, G.; Parodi, G.; Ganau, A. Cardiac Abnormalities in Alzheimer Disease: Clinical Relevance Beyond Pathophysiological Rationale and Instrumental Findings? JACC Heart Fail. 2019, 7, 121–128.
- Gianni, D.; Li, A.; Tesco, G.; McKay, K.M.; Moore, J.; Raygor, K.; Rota, M.; Gwathmey, J.K.; Dec, G.W.; Aretz, T.; et al. Protein Aggregates and Novel Presenilin Gene Variants in Idiopathic Dilated Cardiomyopathy. Circulation 2010, 121, 1216–1226, .
- Subramanian, K.; Gianni, D.; Balla, C.; Assenza, G.E.; Joshi, M.; Semigran, M.J.; Macgillivray, T.E.; van Eyk, J.E.; Agnetti, G.; Paolocci, N.; et al. Cofilin-2 phosphorylation and sequestration in myocardial aggregates: Novel pathogenetic mechanisms for idiopathic dilated cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1199–1214.
- Li, D.; Parks, S.B.; Kushner, J.D.; Nauman, D.; Burgess, D.; Ludwigsen, S.; Partain, J.; Nixon, R.R.; Allen, C.N.; Irwin, R.P.; et al. Mutations of Presenilin Genes in Dilated Cardiomyopathy and Heart Failure. Am. J. Hum. Genet. 2006, 79, 1030–1039, .
- Backs, D.; Saglam, I.; Löffler, C.; Ihne, S.; Morbach, C.; Brenner, S.; Angermann, C.; Ertl, G.; Frantz, S.; Störk, S.; et al. Prevalence of cardiovascular risk factors and diseases in patients with multiple myeloma undergoing autologous peripheral blood stem cell transplantation. Oncotarget 2019, 10, 3154–3165, .
- CQuarta, C.; Kruger, J.L.; Falk, R.H. Cardiac amyloidosis. Circulation 2012, 126, e178–e182.


