1. Introduction: Alzheimer’s—A Systemic Disease?
Alzheimer´s disease (AD) is a neurodegenerative disorder characterized by neurovascular dysfunction
[1], cognitive decline
[2], accumulation of amyloid β peptide (Aβ) in the brain
[3] and the formation of tau-related lesions in neurons called neurofibrillary tangles (NFTs)
[4]. AD is the most common cause of dementia in the elderly and is estimated to affect approximately 14 million people in the United States by 2050
[5]. Different theories about the etiology and pathophysiology of AD have been advanced
[6]. In the amyloid hypothesis, Aβ plaques are causally involved in cognitive decline
[7][8][7,8], and elevated levels of Aβ in brain parenchyma may induce or promote neurovascular
[9][10][9,10] and neuronal dysfunction
[11]. This can lead to a cycle of self-propagation
[12][13][14][12,13,14], as in a prion disease
[15] and ultimately to cerebral β-amyloidosis
[16]. The tau hypothesis postulates that abnormalities in the microtubule-associated tau protein initiate the disease by forming NFTs
[17]. The neurovascular or “two-hit” hypothesis
[18][19][20][18,19,20] claims that two independent pathophysiological events contribute to disease manifestation. First, a cerebrovascular event (hit 1) compromises the blood–brain barrier (BBB), which is critical for brain Aβ homeostasis
[18], and then cerebral Aβ accumulation constitutes the second insult (hit 2). Furthermore, many of the currently available drug therapies are based on the cholinergic hypothesis, in which AD is said to be caused by cholinergic effects such as the reduced synthesis of the neurotransmitter acetylcholine
[21][22][23][21,22,23]. Very recent reports put forth yet another theory: AD is triggered by dysfunctional β-amyloid precursor protein (APP) metabolism or presenilin (PS) mutations. The subsequent accumulation of APP C-terminal fragments may then lead to overproduction and reduced clearance of Aβ
[24]. Finally, there are hypotheses that underscore the importance of environmental risk factors such as air pollution, including smoking, and infections.
While AD is the most common cause of dementia in the elderly, age-related hearing loss (ARHL) is the most prevalent sensory deficit, and growing evidence links it with cognitive decline and an increased risk of dementia. Moreover, it has recently been proposed that AD is a risk factor for heart failure, potentially driven by cardiac amyloidosis. In our review, we set out to present AD as a systemic disease potentially driven by ARHL by highlighting four factors: (i) several diseases and risk factors involving systemic metabolic dysfunction have been linked to an increased risk of developing AD; (ii) no cause-and-effect relationship between these aspects is precisely defined; (iii) common components affecting both the CNS and systemic processes add to AD ontogenesis; and (iv) AD likely represents a multifactorial disease displaying both central and peripheral phenomena.
Specifically, we connect hearing loss to AD development and ultimately to cardiac amyloidosis, while presenting evidence that ARHL is a possible biomarker and modifiable risk factor for cognitive decline, cognitive impairment, and dementia. Moreover, we discuss innovative pursuits to inhibit the Aβ aggregation process and deposition in brain and heart more efficiently through targeted neurotrophic factor supplementation to the cochlea to prevent AD and the development of AD-related amyloid cardiomyopathy.
2. The Blood–Brain Barrier
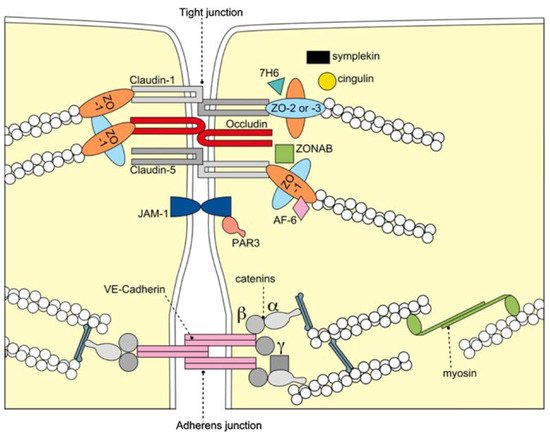
The BBB is formed by the cerebral microvascular endothelium, which tightly controls the homeostasis of the central nervous system (CNS). It consists of endothelial cells (EC), astrocyte end-feet, and pericytes. The basal membrane is responsible for the protection and homeostasis of the brain parenchyma. Chiefly, the tight junctions (TJ) at the zonula occludens in brain capillary ECs create a paracellular barrier that protects the brain from neuroactive substances and other endobiotics and xenobiotics as well as from the external environment. Two different classes of integral membrane proteins constitute the TJ strands in epithelial and endothelial cells: occludin and members of the claudin protein family. In addition, cytoplasmic scaffolding molecules associated with these junctions regulate diverse physiological processes like proliferation, cell polarity and regulated diffusion
[25][26][27][28][29][25,26,27,28,29], (). The BBB is part of the so-called neurovascular unit, which consists of neurons, endotheliocytes, and astrocytes
[30] and connects neurons with the bloodstream. The astrocytes are centrally positioned to mediate interactions between neurons and the cerebral vasculature to maintain BBB integrity and neurovascular coupling. In AD, inflammation within the neurovascular unit promotes the apoptosis of astrocytes, shutting down the neuronal food supply and causing BBB impairment and dysregulation. Moreover, the Aβ impact on the vascular component is defined by Aβ deposition not only in the heart but also in the blood vessels, which leads to the propensity for vessel rupture
[31]. This condition was previously described as cerebral amyloid angiopathy, a common cause of lobar intracerebral hemorrhage and cognitive impairment
[32].
Figure 1. Molecular composition of TJs at the BBB. The transmembrane proteins occludin, the claudin family, and junctional adhesion molecule-1 (JAM-1) constitute the barrier formed by the sealing of the paracellular space by TJs. They appear to interact in a homophilic manner, and occludin seems to co-polymerase into claudin-based TJ strands. BBB = blood brain barrier; TJ = tight junction; VE-cadherin = vascular endothelial cadherin, ZO-1,-2,-3 = zonola occuludens protein-1,-2,-3. Adapted from
[33] with permission.
Inflammation is a central mechanism in AD development
[34]. Under inflammatory conditions and owing to the specialized structure of the BBB, immune cell entry into the CNS parenchyma involves two consecutive, separately regulated steps: migration of immune cells across the BBB or the blood–cerebrospinal fluid barrier (BCSFB) into the CSF-drained spaces of the CNS, followed by progression across the glia limitans into the CNS parenchyma. So far, research has focused mainly on elucidating the distinct molecular mechanisms required for immune cell migration across the different CNS barriers in multiple sclerosis
[35], brain cancer
[36] and ischemic brain injury
[37], while many other neuroinflammatory diseases and comorbidities like AD and AD-related amyloid cardiomyopathy still await a more profound mechanistic investigation.
The inflammation-associated dysfunction of the BBB in AD has been reported to lead to impaired clearance of neurotoxic Aβ, causing cognitive decline
[38][39][38,39]. Cerebrovascular dysfunction appears to be one of the key underlying factors of AD pathogenesis, resulting in cerebral hypoperfusion. Likewise, elevated expression of APP
[40][41][40,41] is thought to result in further Aβ accumulation, predominantly in close proximity to cerebral blood vessels and is detectable in the atherosclerotic intima in 35–60% of patients
[42]. Likely contributors to protracted damage to the cerebral vasculature (also for AD) are the reduced activity of the Pgp-and ATP-binding cassette (ABC) efflux transporters involved in Aβ clearance
[43][44][43,44], altered expression of barrier-constituting TJ proteins
[45], cerebral microbleeds, and vasospasms
[18][19][20][46][47][48][49][50][51][18,19,20,46,47,48,49,50,51].
Accumulating evidence suggests that a mixed vascular pathology and small-vessel disease contribute to the ontogeny of AD
[19][52][19,52]. Moreover, reduced brain–blood perfusion
[53], silent infarcts
[54], and the presence of one or more infarctions
[55] all increase the risk of developing AD, which appears to be caused by various, often modifiable, vascular risk factor affecting BBB integrity
[26].
BBB Dysfunction and Vascular Risk Factors for AD
Epidemiological evidence suggests that cerebrovascular disorders and sporadic late-onset AD carry overlapping common risk factors
[56]. The combination of increased BBB permeability, inflammatory processes and oxidative stress is an acknowledged triad fueling the development of general dementia
[49][57][58][49,57,58]. Generally, diabetes, obesity, hypertension and smoking contribute to inflammation, which fosters the development of AD by promoting BBB dysfunction and increasing its permeability
[59]. Therefore, it might be hypothesized that this situation could trigger Aβ permeation across the BBB into the bloodstream. Additionally, reduced levels of low-density lipoprotein receptors at the BBB contribute to elevated Aβ concentrations in plasma
[60]. As a result, the subsequent Aβ accumulation and deposition occurs in the extracellular region of cardiomyocytes, leading to heart pathologies such as myocardial infarction, cardiomyopathy, and heart failure. Our hypothesis was confirmed by the preliminary findings of Trancone et al., where the authors observed secondary amyloidosis of the heart of AD patients
[61].
Furthermore, hypertension is a key risk factor for developing AD because it induces vascular changes that promote atrophy and increase NFT deposition and Aβ plaque formation
[62][63][64][65][62,63,64,65]. Moreover, mid-life diabetes
[66][67][68][69][66,67,68,69], and obesity
[70][71][72][70,71,72] have been shown to increase the risk for both AD and vascular dementia. Amongst the induced vascular changes, neuroimaging, post-mortem analyses and ultrastructure observations could delineate phenomena such as inflammation-induced basement membrane thickening, reduction or degeneration of pericytes that stabilize the capillaries of the BBB and even the accumulation of erythrocytes, resulting in increased BBB permeability and cognitive decline
[64].
Diabetes: The diagnosis of type-2-diabetes mellitus (T2M) has been positively correlated with an increased risk of developing AD
[67][68][69][67,68,69]. Animal experiments revealed increased and exacerbated cognitive impairment with increased insulin resistance, which is a hallmark of the disease. Increased mitochondrial damage from T2M, thought to amplify the production of cell-toxic ROS, is under investigation as a potential pathophysiological marker
[69][73][69,73].
Obesity: Elevated plasma levels of cholesterol and free fatty acids are suspected not only of promoting a surge in production and deposition of tau and Aβ proteins, but presumably also of pro-inflammatory cytokines. In addition, these might lower the production or expression of matrix metalloproteinases involved in Aβ clearance resulting in BBB dysfunction. A dysfunctional BBB, however, facilitates the accumulation of harmful neurotoxic endo- and xenobiotics, which cause neuronal damage or death, contributing to cognitive decline
[71][72][71,72].
Atherosclerosis in the carotid arteries, which supply blood to the brain, increases the risk of dementias, most prominently AD, through the buildup of plaque in the intimal medial thickness region
[42][43][58][74][75][42,43,58,74,75] ().
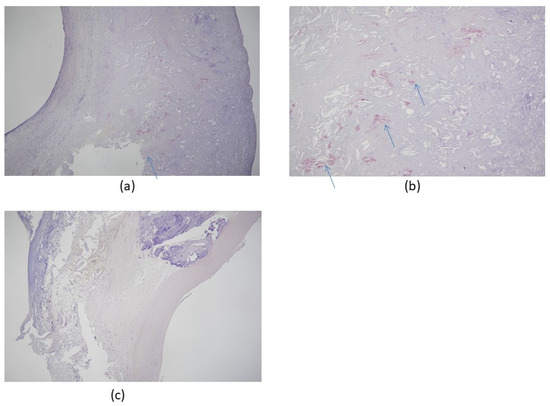
Figure 2. Congo red stain of carotid artery atherosclerosis dissected by carotid endarterectomy. The specimen was fixed in 4% formalin after surgical removal and stained with Congo red for amyloidosis, which colored the Aβ protein plaque deposits red/salmon pink. (a) Congo red stain positive for amyloidosis, 50×; (b) Congo red stain positive for amyloidosis, 100× (hypermagnification of a); (c) negative control, 50×. All patients were scheduled for elective therapeutic endarterectomy and gave informed written consent to the procedure. Arrows indicate amyloid deposits in tissue. Images provided by Hayashi, K., Shimane University Hospital, advanced stroke center.
Morphologically, carotid atherosclerosis is associated with an increased risk of developing dementia
[75] because enhanced intima-media thickening and the formation of plaque promote accelerated and pronounced cognitive decline
[42][43][58][74][42,43,58,74]. gives an example of Congo red staining of amyloid deposits in a typical carotid artery endarterectomy specimen.
Stroke: A significant association between stroke and the development of AD has been acknowledged because it enhances BBB permeability, inflammation and oxidative stress
[49][57][58][49,57,58].
All the abovementioned risk factors contribute to AD development by inducing vascular dysfunction, but for AD and BBB dysfunction, the chicken or the egg dilemma arises.
3. Amyloid Cardiomyopathy
Age-related functional decline in brain and heart function is a typical feature of aging, and a major factor affecting quality of life, psycho-emotional stability, physical strength and self-determination in the elderly
[76]. While AD is the fifth-common cause of death in people older than 65, heart failure is responsible for more than one-third of deaths related to cardiovascular disease
[61][77][61,77]. Heart failure and dementia associated with aging might share common risk factors, mainly related to cardiovascular dysfunction
[61]. If so, it would support the assumption that AD is not limited to brain but is a systemic condition requiring appropriate diagnostic approaches and treatments for both brain and heart disease. Although a causal link has not yet been clearly established, multiple reports describe amyloid cardiomyopathy as a prognostically relevant comorbidity for AD. This raises the possibility of a novel subtype of amyloid cardiomyopathy originating from Aβ inclusions in the myocardial tissue of AD patients. Typically, amyloid cardiomyopathy, a progressive infiltrative condition, is caused by either misfolded monoclonal immunoglobulin light chains (ALs) from an abnormal clonal proliferation of plasma cells that exert direct toxic effects on myocytes or by the abnormal folding of transthyretin (TTR), a liver-synthesized protein (previously called prealbumin) that is involved in the transportation of the hormone thyroxine and retinol-binding protein. The result of the latter is the so-called transthyretin amyloidosis (ATTR). The respective pathomechanisms of ATTR and AL amyloidosis that affect the heart are essentially different
[78]. Common risk factors for the development of amyloid cardiomyopathy seem to include, besides a genetic predisposition, the biochemical effects of ROS formation, inflammation, abnormal coagulation, or silent strokes
[79][80][79,80]. Interestingly, it is well-known that AD patients often develop amyloid cardiomyopathy and peripheral and autonomic neuropathies
[81][82][81,82]. The latter pathology may lead to autonomic cardiac dysfunction/neuropathy that manifests as sinus tachycardia, exercise intolerance, orthostatic hypotension, abnormal blood pressure regulation, dizziness, asymptomatic myocardial ischemia, or infarction
[83][84][83,84].
In the proposed novel AD-related subtype of amyloid cardiomyopathy, the observed HF symptoms are supposed to be the consequence of amyloid deposition in the myocardial tissue. The prevalence of amyloid cardiomyopathy in elderly patients with otherwise unexplained systolic HF or conduction disorders has been estimated at 11% (95% CI 0–23%) and 2% (95% CI 0–4%), respectively.
[85]. In a cross-sectional study, a positive association between an Aβ brain burden measured in vivo and diastolic blood pressure was presented, indicating a possible link between this cardiovascular risk factor and the Aβ burden as measured by Pittsburgh Compound B-positron emission tomography (PiB-PET)
[86]. Amyloid deposits in a typical carotid artery endarterectomy specimen associated with AD are shown in . Carotid endarterectomy is a surgical technique that removes plaque build-up from inside a carotid artery, i.e., from the bloodstream at the neck. It aims to restore normal blood flow to the brain, thereby preventing a stroke. Proof of a direct connection between heart failure and AD is still outstanding, but there are synergistic pathomechanisms between AD and aging because both conditions induce wall thickening and diastolic dysfunction
[79][80][81][82][87][79,80,81,82,87]. Patients with AD were shown to exhibit electrocardiographic and echocardiographic abnormalities, including diastolic dysfunction, which reproduces the pattern of cardiac amyloidosis
[87]. This strengthens the hypothesis that there may be subclinical cardiac involvement in AD that is likely associated with Aβ amyloid deposition
[87]. Gianni et al. first linked cardiac amyloidosis and idiopathic dilated cardiomyopathy (iDCM) by describing a particular pattern of myocardial protein aggregates resembling the tangles and plaques commonly found in AD; however, no overexpression of Aβ was detected
[88]. These findings were corroborated when it was shown that these aggregates closely resembled and were biochemically equivalent to, those found in the brains of AD patients
[89]. Extending these findings even further, genetic variants in the gene associated with early-onset AD (presenilin-1) were identified in familial and sporadic cases of iDCM
[88][90][88,90]. Of note, amyloid in the heart was not limited to AD deposition; that is, it was not secondary to the pathology of another organ; rather, it appeared to be caused by a common underlying mechanism, e.g., systemic light chain AL-amyloidosis
[91] or transthyretin amyloidosis
[61][92][61,92].
A pioneering study that monitored the presence and characteristics of structural and functional cardiac abnormalities in AD patients, was presented by Troncone et al. They investigated the mechanism by which Aβ accumulation in the heart interfered with heart function
[61]. Using imaging and proteomics, they demonstrated that Aβ aggregates can propagate outside the brain, reach the heart and induce AD-related cardiac amyloidosis
[61]. Thus, the toxic effects of Aβ pre-amyloid oligomers (PAOs) on cardiomyocytes were demonstrated
[88], thus causally linking cardiac amyloidosis with AD-related heart failure. This supports the notion that, as with the brains of AD patients, Aβ40 and Aβ42 amyloid plaques and NFTs formed by abnormally hyperphosphorylated microtubules associated with the tau protein constitute a key feature of amyloid cardiomyopathy associated with AD. In this study, Aβ amyloid aggregates were identified both within cardiomyocytes and in interstitial samples from the dysfunctional myocardia of 4 AD patients
[61]. Compared to controls, patients with AD exhibited thicker ventricular walls yet smaller end-diastolic and end-systolic diameters, which were compatible with compromised stroke volume and diastolic function. These observations supported the notion that Aβ amyloid accumulation in the heart contributes to myocardial dysfunction in AD-linked cardiac amyloidosis. In follow-up experiments, the impaired function of cardiomyocytes was demonstrated experimentally and confirmed in AD patients. A peculiar involvement of the heart in AD became apparent, characterized by a generally mild increase in interventricular septum thickness, a higher degree of diastolic dysfunction, and ECG abnormalities, including reduced QRS voltage or voltage/mass ratio
[87]. Consistently, cardiomyocytes of affected patients displayed relaxation deficits, which likely originated from extracellular Aβ infiltrates. In summary, Aβ aggregates were identified both within cardiomyocytes and in the myocardial interstitium of AD patients, proving that Aβ oligomers derived from brain-originating protein misfolding can reach, and may cause damage to, the heart
[3][87][92][3,87,92]. It has, however, not been unraveled yet how Aβ may cross the BBB to induce systemic forms of amyloidosis like cardiac amyloidosis.
Although this data suggest that protein misfolding may affect both the brain and the heart in AD patients, the clinical relevance of this finding still needs to be demonstrated as the sample size of respective clinical studies was very small. Hence, clinicians should attentively monitor cardiac abnormalities when evaluating patients with AD to clarify the full spectrum of myocardial involvement and its comprehensive clinical relevance. Furthermore, the question whether these conditions are part of a common multi-organ syndrome or are causally linked to disparate conditions remains to be answered. Current evidence suggests that AD and HF may be both dependent and independent of each other, but the coexistence of AD and HF points to a new sub-class of amyloid cardiomyopathy with a potentially large impact on quality of life expectancy.