 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Maria Xilouri | + 6276 word(s) | 6276 | 2021-05-11 08:29:39 | | | |
| 2 | Nora Tang | Meta information modification | 6276 | 2021-06-11 04:03:48 | | | | |
| 3 | Nora Tang | Meta information modification | 6276 | 2021-06-17 10:51:14 | | |
Video Upload Options
Accumulation of the neuronal presynaptic protein alpha-synuclein within proteinaceous inclusions represents the key histophathological hallmark of a spectrum of neurodegenerative disorders, referred to by the umbrella term a-synucleinopathies. Even though alpha-synuclein is expressed predominantly in neurons, pathological aggregates of the protein are also found in the glial cells of the brain. In Parkinson’s disease and dementia with Lewy bodies, alpha-synuclein accumulates mainly in neurons forming the Lewy bodies and Lewy neurites, whereas in multiple system atrophy, the protein aggregates mostly in the glial cytoplasmic inclusions within oligodendrocytes. In addition, astrogliosis and microgliosis are found in the synucleinopathy brains, whereas both astrocytes and microglia internalize alpha-synuclein and contribute to the spread of pathology. The mechanisms underlying the pathological accumulation of alpha-synuclein in glial cells that under physiological conditions express low to non-detectable levels of the protein are an area of intense research. Undoubtedly, the presence of aggregated alpha-synuclein can disrupt glial function in general and can contribute to neurodegeneration through numerous pathways.
1. A Role at the Synapse
alpha-Synuclein (aSyn) is a small, intrinsically disordered protein that is mainly localized at the pre-synaptic terminal [1][2], but is also present in the neuronal somato-dendritic compartment [3], in red blood cells [4], in the gut and other peripheral tissues [5][6][7]. Although aSyn is highly enriched in presynaptic boutons, it displays a delayed distribution in the terminals, suggesting that it is implicated in later stages of synaptic development, rather than playing a central role in synapse modulation [2]. Importantly, aSyn is differentially expressed in the various neuronal cell types, being more abundant in excitatory synapses across different brain regions and particularly in central catecholaminergic systems [8]. On the contrary, the protein displays a differential expression profile in inhibitory synapses amongst the different brain areas, with a particular interest of aSyn presence in striatal GABAergic medium spiny neurons (MSNs) [9][10].
The first indication regarding the role of aSyn on neural plasticity arose about 25 years ago, when “synelfin” (synuclein, NACP) expression was found up-regulated during bird song learning [11]. The localization of aSyn in pre-synaptic boutons is mainly attributed to its tight association with synaptic vesicle membranes [12] and its high affinity for the SNARE complex proteins synaptobrevin-2 (or Vesicle Associated Membrane Protein 2, VAMP2), synapsin III and rab3A [13][14][15]. It has been proposed that aSyn interacts with VAMP2 and promotes SNARE complex assembly [13], followed then by its disassembly in order to complete the round of membrane fusion (Figure 1). The crucial role of aSyn assembly with SNARE complex on neuronal survival was further verified by the neuronal dysfunction and impaired survival of triple αβγ-synuclein knockout mice during ageing [13][16]. Interestingly, aSyn lentiviral overexpression in primary neurons led to enhanced SNARE complex assembly, further supporting the role of this protein in synaptic activity [13]. The same group later showed that only multimeric membrane-bound, but not the soluble monomeric aSyn, can promote the SNARE complex assembly [17]. It has been also recently suggested that aSyn is involved in synaptic vesicle homeostasis at the pre-synaptic terminal via a calcium (Ca2+)-dependent mechanism [18].
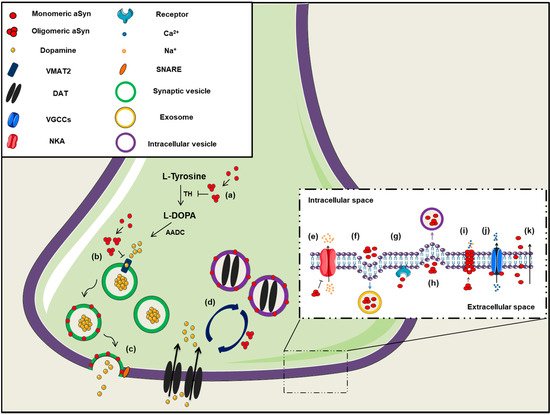
Figure 1. The role of aSyn at the presynaptic terminal. A schematic representation depicting of aSyn physiological and pathological effects at the synapse: (a) aSyn reduces the activity of tyrosine hydroxylase (TH), the enzyme responsible for catalyzing the conversion of L-Tyrosine to L-DOPA, thus impairing dopamine biosynthesis, (b) Increased levels of aSyn inhibit VMAT2, which is responsible for the uptake of monoamines (such as dopamine) into synaptic vesicles, (c) aSyn associates with synaptic vesicle membranes and regulates the SNARE-dependent vesicle fusion and neurotransmitter release, (d) Soluble aSyn interacts with the dopaminergic transporter DAT and decreases its amount on the plasma membrane, thus regulating the dopamine re-uptake from the synapse. However, aSyn aggregates trigger DAT recruitment to the plasma membrane that leads to massive entry of dopamine, (e) aSyn aggregates interact with Na+/K+-ATPase (NKA) preventing the effective pump out of Na+ ions, (f) aSyn is secreted from neuronal cells partly via associating with exosomes, (g) Extracellular aSyn interacts with neuronal receptors (i.e., LAG3) for its internalization in neurons or (h) it is up-taken via endocytosis, (i) PD-linked A30P and A53T mutant aSyn form large membrane pores through which most cations (i.e., Ca2+) can pass non-selectively, (j) Extracellular aSyn activates the voltage-gated Ca2+ channels (VGCCs), resulting in increased Ca2+ influx, (k) Monomeric aSyn enters neuronal cells via passive diffusion or direct penetration of their plasma membrane.
On the contrary, natively unfolded monomeric aSyn at the pre-synaptic terminal is prone to form pathological conformations, thus exerting neurotoxic effects [19] (Figure 1). It has been additionally suggested that aSyn is preferably bound to synapsin 1 and VAMP2 when the protein is present in its oligomeric form [20], highlighting the importance of the conformational state of aSyn for its proper function. There are also findings supporting the implication of aSyn in synaptic transmission, due to its association with the synaptic vesicle pool, modulating the vesicle mobility, the recycling pool homeostasis and endocytosis [21][22][23].
Alpha-synuclein can also function as a molecular chaperone via effective binding to other intracellular proteins. The first indication came with the discovery that aSyn displays structural and functional homology with other molecular chaperones, as the 14-3-3 or small heat shock proteins [24][25]. Additional studies revealed that aSyn synergistically acts with the presynaptic cysteine-string protein-alpha (CSPalpha) promoting the assembly of the SNARE complex [13][26], further validating its chaperoning properties. Biochemical and structural analysis of aSyn strengthened the current indications for its chaperone-like function via its C-terminal region (residues 61-140) [27][28][29]. However, following studies indicated that the chaperone-binding site of aSyn lies within the non-amyloidal component (NAC) region (residues 61-95), which is prone to aggregation and thus highly susceptible to form fibrils [30][31].
2. Association with Membranes and Lipid Trafficking
Intracellular aSyn can be found either natively unfolded in a soluble state or membrane-bound forming an alpha-helical or a beta-sheet secondary structure, depending on the solution conditions [32][33][34]. It has been proposed that there is a bidirectional link between aSyn species formation and membrane remodeling, meaning that not only aSyn structure is affected upon lipid interaction, but also that membrane integrity depends on the presence of different aSyn conformations [35][36][37]. However, there are controversial results regarding the association of aSyn with membrane lipids and its conformational state, with some studies reporting that membrane-bound aSyn gets protected from aggregation, thus leading to neurotoxicity attenuation [19][38][39], whereas others suggest that interaction of aSyn with membranes triggers its self-association and subsequent aggregation [40][41][42]. Importantly, it has been shown that the PD-related aSyn mutations reduce its interaction with membranes, thus further suggesting that aSyn binding on membranes may exert neuroprotective effects [43][44][45][46][47].
A plethora of studies argue that aSyn in its soluble state exists as a monomer [48][49][50][51], whereas others suggest that it occurs physiologically as a tetramer resisting aggregation [52][53][54]. In the presence of lipid membranes, aSyn adopts an alpha helical structure in the N-terminus region that stabilizes the formation of high-order aSyn multimers [17][48][55][56]. Interestingly, the membrane curvature seems to affect the structure of aSyn, which can adopt either an elongated or a broken alpha-helix conformation, when bound to a large diameter (∼100 nm) or a small, highly curved vesicle, respectively [57][58][59][60]. It has been also proposed that aSyn has a role in lipid metabolism, since it participates in fatty acids transportation between the cytosol and membranous compartments [61][62] and in lipid and membrane biogenesis organizing and stabilizing the lipid bilayer of membranes and vesicles [63][64]. On the other hand, disrupted aSyn expression pattern leads to lipid dysregulation, since both the absence and the overexpression of either wild-type (WT) or mutated aSyn gives rise to abnormal lipid metabolism [65][66][67][68]. Finally, several studies have demonstrated that aSyn regulates membrane homeostasis via inhibition of phospholipases activity, such as phospholipase D [69][70][71][72]; however, there are controversial results in the literature [73].
3. Aggregation and Post-Translational Modifications
alpha-Synuclein is composed of three distinct domains: the N-terminal lipid-binding domain, the NAC region and the C-terminal binding domain [59][74][75]. A central role in the fibril formation and subsequent aggregation of aSyn is thought to be mediated through the NAC region of the protein composed of nonpolar side-chains and assembles cross b-structures. Based on that, it has been shown that the deletion of specific residues (74-84) within the core region can abolish aSyn aggregation [76][77]. It has been also demonstrated that the endogenous neuronal aSyn and the interaction of aSyn with lipids plays a central role for aSyn recruitment and subsequent seeding of pathology, as it could behave as a core for the formation of insoluble aggregates [10][50][78][79].
Several mutations in the Snca gene have been linked to PD pathogenesis, such as the A53T, A30P, E46K, H50Q, G51D, A18P, pA29S and A53E mutations, all located in the N-terminus region [80][81][82][43][83][84][85]. Most of them are tightly linked to enhanced aSyn aggregation, pathology progression and clinical manifestations in PD. Specifically, A53T and A30P aSyn mutants are natively unfolded, similarly to WT protein. However, at higher concentrations A53T has been shown to accelerate aSyn fibrillization, a critical event in PD pathogenesis [86][87][88]. On the other hand, A30P promotes aSyn oligomerization rather than fibrillization, thus reducing aggregate formation [87][89]. The E46K mutation leads to conformational changes of aSyn due to C-terminal to N-terminal contacts in the monomeric protein, resulting in enhanced aSyn accumulation [89][90][91]. Moreover, the PD-linked H50Q point mutation increases aSyn aggregation propensity and toxicity [92], whereas the G51D mutation has the opposite effects [93]. However, although G51D mutants seem to oligomerize in a slow rate, they form more toxic fibrils, thus suggesting distinct disease mechanisms for the various aSyn mutations [94][95]. Similarly, A53E mutant seems to lead to neuronal toxicity via an aSyn aggregation-independent manner [96]. Strikingly, the G51D and A53E aSyn mutations have been proposed as potential links between PD and MSA [84][97]. However, up-to-date, no hereditable mutations in the coding region of SNCA gene have been identified in MSA cases [98]. Apart from point mutations [95][99][100], various post-translational modifications are implicated in aSyn aggregation, the most important of which are phosphorylation, sumoylation, ubiquitination, nitration, N-acetylation, O-GlcNAcylation and truncation.
The phosphorylation of aSyn both at serine and tyrosine residues and particularly at Ser129 is widely considered as an indicator of pathology. However, the effect of Ser129 phosphorylation on aSyn toxicity is still under debate, with the majority of studies suggesting that it accelerates cell toxicity and neurodegeneration [101][102][103][104][105]. Contrarily, others have proposed a neuroprotective role of Ser129 phosphorylation since it was reported to drive the conversion of toxic oligomers into less harmful aggregates [106][107][108]. Other mechanisms of phosphorylated Ser129 aSyn-mediated neuroprotection include inhibition of its fibrillation [109], upregulation of tyrosine hydroxylase (TH) activity [110] or lowering of the protein’s membrane-binding affinity [111]. Although the 90% of aSyn in LBs is found phosphorylated at Ser129, a significant amount of phosphorylated Ser129 aSyn is also detected in a soluble, rather than in an aggregated state in PD brains [112], whereas only a small percentage of aSyn is phosphorylated at Ser129 in the brains of healthy controls [113][114][115]. In addition, aSyn can be phosphorylated at Ser87, Tyr125, Tyr133 and Tyr136 residues [116][117] and these are also implicated in either neurotoxic or neuroprotective events [105][116][118][119]. Nonetheless, in most in vivo models where aSyn is overexpressed (virally, transgenic or PFF-inoculations) the detection of pSer129 positive aSyn signal is invariably linked to neurotoxicity, indicating a rather neurotoxic and not a neuroprotective role.
Nitrated aSyn is also tightly linked to neurodegeneration, as demonstrated by experiments in both cellular and animal models, as well as in patient-derived brains [120][121][122][123], through its implication in oxidative damage and disease development [124]. Four tyrosine residues in aSyn sequence, Tyr39 (within the N-terminus), Tyr125, Tyr133 and Tyr 136 (within the C-terminus) can undergo nitration. Nitration at Tyr39 has been shown to result in low binding affinity of aSyn on lipid vesicles due to its loss-of-alpha helical conformation status [125], whereas nitration at Tyr125 seems to play a crucial role for aSyn dimerization [126]. Moreover, the linking between two tyrosines is considered as a potential mechanism for aSyn oligomer stabilization and its subsequent aggregation into proteinaceous inclusions [127]. In addition, the detection of nitrated aSyn in the human blood serum could potentially serve as a clinical biomarker for PD diagnosis [128].
Another aSyn post-translational modification crucial for its aggregation propensity is ubiquitination, via regulation of the proteasome-dependent protein degradation [129] and the subcellular localization of the protein [130]. Ubiquitinated aSyn has been isolated from LBs and sarkosyl-insoluble fractions derived from synucleinopathy brains [131][132]. CHIP (C-terminal U-box domain of co-chaperone Hsp70-interacting protein), SIAH (seven in absentia homolog) and Nedd4 (neuronal precursor cell-expressed, developmentally down-regulated gene 4) have been identified among the E3 ubiquitin ligases implicated in aSyn ubiquitination [133][134][135][136][137][138]. Ubiquitin modification has been demonstrated to have differential effects on aSyn accumulation and subsequent aggregation, dependent on the residue being modified. More precisely, ubiquitination at Lys6, Lys12 and Lys21 residues has been shown to moderately inhibit aSyn fibrillation, whereas at Lys10 and Lys23 residues has been reported to promote the formation of aSyn inclusions [139]. In addition, ubiquitination at Lys32, Lys34, Lys43 and Lys96 inhibits aSyn aggregation [139].
Sumoylation is a similar process to ubiquitination, since aSyn is conjugated to SUMO (small ubiquitin-like modifier) at lysine residues. SUMO-1 was found in aSyn-positive inclusions of a-synucleinopathy brains or associated with lysosomes of PD animal models [140][141][142]. It has been also suggested that aSyn sumoylation facilitates its aggregation since it inhibits its degradation [143], whereas other studies proposed a neuroprotective role of aSyn sumoylation, which seems to promote aSyn solubility and thus inhibit its aggregation [144][145]. The discrepancy between these data may be attributed to the different lysine residues available for sumoylation being investigated in each study. Another aSyn modification that has been up for debate is its N-terminal acetylation. Although many studies have assigned a neurotoxic role on aSyn N-acetylation, as it has been shown to promote aSyn β-sheet formation and fibrillation [146][147][148], others suggest that either N-acetylated aSyn mediates its physiological binding on synaptic vesicles [149], or it acts in a protective manner against aSyn aggregation [150][151].
O-GlcNAcylation is a biochemical process that involves the attachment of O-linked N-acetylglucosamine to Ser and Thr residues of various proteins, amongst which is aSyn. Murine and human aSyn have been shown to be O-GlcNAcyled in many threonine residues including Thr33, Thr34, Thr54, Thr59, Thr64, Thr72, Thr75, Thr81 and Thr87 [152][153][154][155][156] and this post-translational modification has repetitively been linked to reduced aSyn aggregation and attenuation of PD-related toxicity [157][158][159][160]. Finally, aSyn truncation has gained scientific attention, given that C-terminally truncated aSyn has been identified in the inclusions present in PD brains [161][162][163]. Many studies have considered that aSyn truncations have neurotoxic effects due to increased accumulation of misfolded aSyn [164][165][166][167][168][169][170][171][172].
4. Channel Formation/Channel Interactions
As mentioned above, membrane-bound aSyn adopts an alpha-helical conformation, which facilitates its oligomerization and subsequent aggregation. It has been suggested that aSyn oligomers can form transmembrane channels and pore-like structures that have been linked to pathological events during PD development (Figure 1) [173][174][175]. As a result, vesicles or low-molecular mass molecules may penetrate the cell membrane and in combination with altered cellular ionic homeostasis could potentially lead to cell toxicity and neuronal degeneration [176][177]. Another mechanism for the increased membrane permeability involves the incorporation of aSyn oligomers between the membrane phospholipids, thus leading to the bilayer thinning which thereafter allows the diffusion of small molecules [178].
A wide range of studies has demonstrated that the ion channels formed by oligomeric aSyn dysregulate cellular ion concentrations and may represent a critical event in the pathogenesis of a-synucleinopathies [176]. Some PD-linked aSyn mutations, such as E46K and A53T, have been shown to be implicated in the channel formation, whereas other aSyn mutants (i.e., A30P) have displayed low membrane affinity [175][179]. However, other groups have shown that A30P and A53T aSyn mutations are responsible for the formation of large membrane pores through which most cations can pass non-selectively [180]. It has been reported that the formation of such cation-permeable pores could lead either to ion conductivity or to increased Ca2+ influx and subsequent cell death [180][181][182][183]. Upon aSyn cation channel opening, other channels, such as the ATP-dependent potassium channels K (ATP), have been reported to be activated in hippocampal neurons and this could probably diminish the aSyn-dependent neuronal excitability [183].
Binding of aSyn to the plasma membrane results in the formation of aggregates and this aggregation leads to the redistribution of the α3 subunit of Na+/K+-ATPase. As a result, Na+/K+-ATPase is no longer able to effectively pump out Na+ from neurons, thus leading to an intracellular Na+ accumulation [184]. Furthermore, extracellular aSyn was reported to activate the voltage-gated Ca2+ channel Cav2.2 in rat neurons, due to disorganization of lipid rafts in the plasma membrane, resulting in enhanced dopamine release and increased Ca2+ influx [185]. Both events may explain the synaptic dysfunction and neuronal vulnerability in PD. L-type Ca2+ channels are also implicated in PD development, as administration of L-type Ca2+ channel blockers (i.e., isradipine, nimodipine) in animal models and PD patients, reduced death risk and ameliorated disease manifestations [186][187][188][189]. Finally, aSyn oligomers can inhibit α4β2 nicotinic acetylcholine receptors of dopaminergic neurons, thus leading to cholinergic signaling deficits [190]. In summary, aSyn seems to regulate neuronal toxicity and survival via the formation of channels or pores in the plasma membrane or via its interaction with other channels or receptors crucial for the proper neuronal activity (Figure 1).
5. Dopamine Metabolism
Soluble aSyn has been proposed to interact with the dopamine transporter (DAT) and decrease its amount on the plasma membrane, thus regulating the dopamine re-uptake from the synapse and protect neuronal cells from excessive dopamine toxicity [191][192]. Contrariwise, aSyn aggregation triggers DAT recruitment to the plasma membrane that results in massive entry of dopamine and production of reactive oxygen species (ROS) in neurons [193]. It is obvious that aSyn-mediated modulation of DAT activity is crucial for neuronal functioning via a balanced dopaminergic neurotransmission. Moreover, the regulation of dopamine storage is provided by an interaction of aSyn with the vesicular monoamine transporter 2 (VMAT2), which is responsible for the packaging of monoamine transmitters into synaptic vesicles [194]. It has been reported that increased levels of aSyn lead to VMAT2 inhibition and dopamine dysregulation that results in pathological events [195]. In addition, aSyn regulates dopamine biosynthesis, via reducing the activity or the phosphorylation status of TH, the rate-limiting enzyme in catecholamine synthesis [196][197][198][199][200][201]. In agreement, enhanced expression or phosphorylation and subsequent aggregation of aSyn alter TH activity and evoke an imbalance in dopamine synthesis, thus leading to neurotoxicity [110][202][203][204]. In vivo evidence further support the role of aSyn in dopamine metabolism, since the absence of aSyn caused decreased reuptake of dopamine, low levels of TH and DAT in the mouse striatum and reduced number of dopaminergic cells in the substantia nigra of aSyn KO mice [205][206][207].
6. Interaction with Mitochondria and ER
alpha-Synuclein displays a remarkable conformational flexibility upon macromolecular interactions and can associate with mitochondrial membranes, thus altering mitochondrial function [208][209][210] (Figure 2). There are reports suggesting that aSyn is a physiological regulator of mitochondrial activity [211][212][213], whereas others support the opposite [214][215][216]. Such discrepancies could be attributed to the different synuclein models utilized in each study, taking into account that brain homeostasis is a complex process and in vivo studies recapitulate better the interplay between the various brain components, compared to the isolated in vitro cellular setup. A bidirectional interaction between aSyn aggregation and mitochondrial dysfunction has been implicated in PD pathogenesis. In particular, increased levels of aSyn can lead to mitochondrial dysfunction [217][218][219][220][221][222], whereas, conversely, impairment of mitochondrial activity may accelerate aSyn pathology [223][224][225][226]; however, the precise underlying mechanisms remain to be elucidated. Both WT and mutant aSyn have been shown to interact with mitochondrial elements, altering both mitochondria morphology and function. Specifically, soluble pre-fibrillar aSyn oligomers seem to be responsible for complex I dysfunction, loss of membrane potential, disrupted Ca2+ homeostasis, enhanced cytochrome c release and ROS production, thus leading to neuronal demise [218][227][228][229][230].
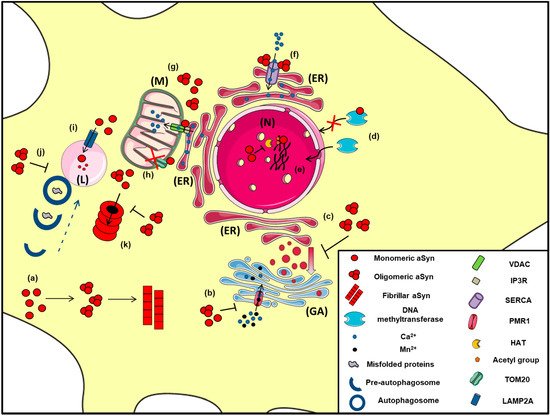
Figure 2. The proposed intracellular effects of various aSyn conformations in neurons. A schematic representation of the aberrant interactions between the various aSyn species with intracellular organelles: (a) In the cytoplasm of neurons, aSyn monomers form oligomers that can eventually become fibrils, (b) Both unfolded and aggregated aSyn impair the function of PMR1, a Ca2+-transporting ATPase pump that regulates Ca2+ and Mn+2 levels in the Golgi apparatus (GA), resulting in elevated cytosolic Ca2+ levels, (c) Both WT and mutant A53T aSyn disrupt the vesicular transport from Endoplasmic Reticulum (ER) to Golgi (GA), (d) WT aSyn inhibits the transportation of methyltransferases from the cytoplasm to the nucleus (N), thus altering DNA methylation of the SNCA gene, (e) Inside the nucleus (N), aSyn inhibits histone acetylation via its direct binding to histones or by preventing the action of histone acetyltransferase (HAT) enzymes, thus interfering in the process of gene transcription, (f) In the ER, aSyn aggregates activate the Ca2+-ATPase SERCA, resulting in dysregulated Ca2+ homeostasis, (g) Both monomeric and oligomeric aSyn interact with Voltage-dependent anion channel 1 (VDAC1) and inositol triphosphate receptors (IP3Rs), the protein components involved in mitochondrial-associated ER membrane (MAM) and regulates the transmission of Ca2+ signals from the ER to mitochondria (M), (h) aSyn binds to TOM20, a mitochondrial import receptor subunit and inhibits normal protein import, (i) Normally, monomeric or dimeric forms of aSyn are degraded in the lysosome (L) via Chaperone Mediated Autophagy (CMA), following their interaction with LAMP2A. However, under pathological conditions, impairment of CMA has been proposed to lead to aSyn accumulation and subsequent cell toxicity, (j) Oligomeric aSyn and various misfolded proteins are cleared via macroautophagy, following the fusion of autophagosomes with the lysosome. Pathological aSyn has been shown to inhibit autophagosome maturation or their fusion with lysosomes, thus impairing autophagic flux, (k) Monomeric and oligomeric aSyn are degraded via the proteasome; however, under pathological conditions, increased levels of aSyn or even soluble aSyn oligomers may inhibit proteasomal function, leading to aSyn accumulation and the formation of insoluble aggregates.
Experiments in various animal models of a-synucleinopathy have revealed mitochondrial abnormalities, DNA damage and neuronal degeneration in PD-affected brain regions [222][231][232]. Moreover, in vitro and in vivo experiments have shown that aSyn inhibits mitochondrial fusion and triggers mitochondrial fragmentation [209][233]. Di Maio and colleagues have proposed that certain post-translationally modified aSyn conformations (soluble oligomers, dopamine-modified and S129E phosphorylation mimic) lead to impaired mitochondrial function via binding to TOM20 (translocase of the outer membrane receptor) and inhibiting mitochondrial protein import [217].
Nonetheless, there is evidence suggesting an impairment of mitochondrial function upstream of aSyn pathology. Experiments using the pesticides rotenone and paraquat have shown that dysregulation of mitochondrial function leads to nigrostriatal dopaminergic loss and formation of LB-like inclusions, positively stained with anti-aSyn antibodies and thioflavine S, thus resembling PD features [224][225][234][235][236]. Similarly, incubation of WT aSyn-overexpressing COS-7 cells with mitochondrial inhibitors resulted in the disappearance of the aSyn aggregates formed upon rotenone or oligomycin treatment [237]. A plethora of studies that utilize the mitochondrial neurotoxin MPTP to induce PD-like pathology in animals, further suggest that mitochondria impairment is a key player in disease development [223][226][238][239][240][241][242]. Genetic studies further support the hypothesis of aSyn accumulation as a secondary event following mitochondrial malfunction. Specifically, mutations in ATP13A2 (ATPase cation transporting 13A2), encoding for the lysosomal type 5 P-type ATPase, were shown to result in dysregulation in mitochondrial depolarization and ATP metabolism leading to mitochondrial fragmentation and subsequent cell death [243][244].
Apart from its implication in mitochondrial failure, aSyn has been also reported to play a biological role in the association of mitochondria with the endoplasmic reticulum (ER) Ca2+ homeostasis. It has been demonstrated that aSyn favors the Ca2+ transfer from ER to mitochondria, as a result of the communication the two organelles, probably due to the fact that aSyn can act as a “bridge” via its C terminus [245]. Later studies further supported the physiological localization of aSyn in mitochondria-associated ER membranes (MAM), stabilizing their interaction, which was perturbed upon aSyn aggregation and its subsequent redistribution [246][247]. Interestingly, the familial PD-linked A53T and A30P aSyn point mutations resulted in their weakened interaction with MAM, which affected MAM function and mitochondrial integrity [247].
The association of aSyn with mitochondria was further corroborated by findings indicating interactions between both monomeric and oligomeric aSyn with the Ca2+ transporting voltage-dependent anion channel 1 (VDAC1) [248][249][250][251]. Importantly, VDAC1 has been detected on the MAM of ER mediating the communication between the two organelles, regulating Ca2+ homeostasis [252][253][254]. Moreover, VDAC levels have been found decreased in nigral neurons of PD brains, where pathological aSyn inclusions had been formed [255]. Additionally, VDAC has been proposed to be a component of the mitochondrial permeability transition pore, the opening of which has been shown to be affected by aSyn overexpression and oligomerization [208][256]. In vivo experiments on transgenic mice overexpressing the human A53T aSyn further supported the role of permeability transition pore activity modulation on the mitochondrial dysfunction during PD pathogenesis [257].
7. Unfolded Protein Response, Regulation of ER/Golgi Trafficking and Ca2+ Homeostasis
The ER is a continuous membrane system mainly responsible for the production and processing of lipids and proteins, as well as Ca2+ homeostasis. In case of impaired protein folding (ER stress), cells activate a group of signal transduction pathways, known as the unfolded protein response (UPR). It has been previously shown that aSyn overexpression in PD patients leads to UPR and contributes to the molecular pathogenesis of the disease [258]. The ER chaperone glucose regulated protein 78 (GRP78/BIP) has a crucial role on ER stress regulation due to its ability to control the activation of transmembrane ER stress sensors (IRE1, PERK and ATF6) [259]. Disassociation of GRP78 from IRE1 and PERK results in stress signaling, finally leading to altered ER homeostasis [260]. aSyn associates with GRP78/BIP under physiological or pathological conditions, thus inducing UPR and leading to dopaminergic cell death [20][261]. Strikingly, Ser129 phosphorylated and aggregated aSyn was found in ER microsomes of A53T transgenic mice and more importantly, administration of the UPR inhibitor salubrinal, effectively attenuated disease manifestations in this PD-mouse model [262][263]. It is worth mentioning that GRP78/BiP levels were found elevated in DLB and PD brains in an aSyn burden-dependent manner [264]. In addition, the protein levels of various ER chaperones were found elevated in a-synucleinopathy models, co-localized with aSyn positive inclusions, suggesting that aggregated aSyn could potentially be implicated in UPR regulation in disease progression [262][265][266][267][268][269][270][271].
Proteins synthesized in the ER, are packaged into vesicles and directed to Golgi apparatus for subsequent modifications. One of the first pathological roles attributed to aSyn is the blockade of the vesicular transport from ER to Golgi by antagonizing ER/Golgi SNAREs [272][273][274]. Towards the same direction, aSyn can also disrupt the intra-Golgi and post-Golgi secretory trafficking, via an abnormal interaction with several Rab-family proteins of the intracellular endocytic pathway [272][274][275][276][277]. Additionally, aSyn can also impair the ionic transport and membrane trafficking, resulting in Golgi fragmentation and subsequent cytotoxicity [278][279][280].
Another significant role of aSyn on ER and Golgi function is the regulation of Ca2+ homeostasis via its binding on specific channels or pumps localized in these organelles (Figure 2). Specifically, proximity ligation assay experiments demonstrated that soluble and insoluble aSyn aggregates, but not monomers, interact with the ER Ca2+-ATPase SERCA, resulting in decreased cytosolic Ca2+ that disrupts the physiological cell function and leads to neuronal cell death [281]. Moreover, administration of the SERCA inhibitor cyclopiazonic acid restored cytosolic Ca2+ levels and protected neurons against the aggregated aSyn-dependent cell death [281]. In support to these results, aggregated aSyn bound on SERCA pump was detected in LBs and GCIs of PD and MSA brains, respectively [281]. Furthermore, PMR1, a Ca2+-transporting ATPase 1 pump regulating the levels of Ca2+ and Mn+2 ions in the Golgi [282], has been proposed to be a mediator of aSyn-dependent cytotoxicity. Specifically, in various PD models (yeast, flies and nematodes), PMR1 pump has been linked to aSyn pathology via a Ca2+-dependent mechanism, where aSyn accumulation elevated cytosolic Ca2+ levels and increased cell death. Interestingly, upon PMR1 deletion, the disease-associated characteristics were abolished, further suggesting the relevance of this pump to aSyn pathology [283][284].
8. a-Synuclein in the Nucleus
The name aSyn was given to the protein due to its localization in the nucleus and presynaptic nerve terminals [12]. Nuclear aSyn was detected in neurons of various brain regions of rodents and was reported to interact with histones, underlying PD pathology [285][286][287], even though a single study declares that the nuclear staining of aSyn is attributed to the non-specific signal of some antibodies that probably recognize unknown antigens in neuronal nuclei [288]. It has been proposed that aSyn is responsible for epigenetic dysregulation via inhibition of histone acetylation or reduced DNA methylation, thus favoring neuronal degeneration, whereas others suggest that nuclear aSyn regulates cell cycle rate exhibiting cell toxicity [289][290][291]. Importantly, histone deacetylase (HDAC) inhibitors attenuated aSyn toxicity and provided neuroprotection in both cell culture and transgenic Drosophila models [289][292].
Experiments in SH-SY5Y cells revealed that nuclear translocation of aSyn is regulated by calreticulin and Ca2+, following treatment with retinoic acid and modulates the expression of PD-linked genes such as ATP13A2 and PINK1 (PTEN-induced kinase1) [293]. Interestingly, phosphorylated aSyn at Ser129 was found accumulated in the nucleus of HEK293E-aSyn overexpressing cells and in various brain regions of transgenic (Thy1)-[A30P] aSyn mice [294]. Further experiments in H4 cells expressing various aSyn proteins verified that nuclear localization of aSyn depends on its phosphorylation at Ser129 [295]. The same group supported a role of DNA-binding and gene expression regulation for aSyn providing an insight into the role of modified aSyn in the nucleus [295]. Furthermore, other post-translational modifications of aSyn, such as sumoylation, seem to be responsible for the translocation of aSyn from the cytoplasm to the nucleus [296]. Although the majority of studies support a neurotoxic role for aSyn nuclear localization, some groups proposed that aSyn in the nucleus displays a protective role against DNA damage, replication stress or impaired nucleo-cytoplasmic transport [297][298][299]. However, the numerous in vitro and in vivo studies demonstrating a neurotoxic role of nuclear aSyn, in contradiction to the limited number of studies supporting a protective role originated mostly from cell lines or yeast, favors the pathological potential of nuclear aSyn.
9. Alpha-Synuclein and Protein Degradation Pathways: An Intricate Interplay
A great wealth of data focuses on the complicated relationship between aSyn clearance and protein degradation pathways (Figure 2). Both the ubiquitin-proteasome system (UPS) and the autophagy lysosome pathway (ALP) are responsible for aSyn degradation in a manner that depends on cell type, tissue and aSyn conformation state [300][301][302]. Specifically, there are studies demonstrating that aSyn can be degraded by the 26S/20S proteasome via ubiquitin-dependent [303][304] and ubiquitin-independent manner [305][306]. Studies in PC12, HEK293 and primary mesencephalic cells suggested that pharmacological inhibition of the proteasome does not lead to aSyn accumulation [302][307][308]; however, others have shown that soluble aSyn oligomers, but not monomers, are partially cleared via the 26S proteasome [309]. Importantly, it has been proposed that the UPS is responsible for aSyn removal under normal conditions, while in pathological cases the ALP is recruited to clear the increased aSyn burden [310].
Chaperone-mediated autophagy (CMA) is also responsible for the degradation of monomeric or dimeric forms of the protein via the lysosome-associated membrane protein type 2A (LAMP2A), whereas oligomeric aSyn is cleared mainly via macroautophagy [302][311][312]. Lee and colleagues also suggested that the lysosome is responsible for the removal of oligomeric but not fibrillar aSyn and that lysosomal failure results in aSyn accumulation and aggregation and subsequent cell death [313]. Moreover, initial in vivo evidence suggested that increased aSyn protein levels evoked by paraquat treatment were preferably degraded via CMA in dopaminergic neurons, where the levels of LAMP2A and the lysosomal heat shock cognate protein of 70 kDa (HSC70), both essential CMA-components, were found elevated [314]. We have also shown that boosting CMA function via LAMP2A overexpression in cell lines and primary neuronal cultures and in the rat dopaminergic system mitigated aSyn protein levels and related toxicity [315]. Similar neuroprotective effects were obtained upon LAMP2A overexpression in the Drosophila brain [316]. On the contrary, we have also shown that LAMP2A silencing led to endogenous aSyn accumulation in vitro [302] and in vivo [317] and in extensive neurodegeneration of the rat nigrostriatal axis [317]. Decreased levels of LAMP2A and HSC70 were reported in the human substantia nigra and amygdala of PD brains [318], whereas, in a subsequent study, LAMP2A was found to be selectively reduced in association with increased aSyn levels, even in the early stages of PD, thus suggesting a potential dysregulation of CMA-mediated protein degradation prior to substantial aSyn aggregation in PD [319].
However, a bidirectional link between aSyn accumulation and the protein degradation machineries exists and extensive studies have been conducted to elucidate not only the manner of aberrant aSyn degradation in a-synucleinopathies, but also the impact of various aSyn conformations on UPS and ALP function. It has been proposed that overexpression of A30P and A53T mutants, contrarily to WT aSyn, leads to cell death due to proteasomal inhibition [320]. Indeed, overexpression of mutant A53T aSyn resulted in UPS failure by inhibiting the activity of the 20S/26S proteasome, finally leading to aSyn pathological accumulation [321]. Other groups have failed to detect alterations in the proteasomal function of PC12 cells or transgenic mice, following overexpression of WT or mutant (A30P, A53T) aSyn [322]. Moreover, later studies demonstrated that transient overexpression of WT or mutant aSyn, followed by addition of recombinant aSyn oligomers and fibrils in an osteosarcoma cell line, did not result in any disturbance of the proteasomal function [323]. Importantly, studies in human post-mortem PD brains also suggested impaired proteasomal function in the substantia nigra [324][325][326], further supporting a role of UPS malfunction in PD pathogenesis. In addition, total rates of protein degradation declines with aging, thus contributing to the pathogenesis of age-related diseases [327]. Even though human post-mortem studies provide valuable information in regards to etiology and/or disease pathogenesis, the data obtained should be treated with caution, given into account the overall decline in the function of multiple systems with aging. For a-synucleinopathies, we believe that the use of tissue from affected and non-affected (in regards to aSyn pathology and neuronal death) brain areas may provide useful information regarding early or late events leading to neurodegeneration.
Increased aSyn protein burden is reported to impair macroautophagy function as well, via its interaction with Rab1a, an event that subsequently results in the autophagosome-formation-related protein Atg9 mislocalization [328]. Similar results were obtained from cells expressing the PD-linked mutation of the retromer protein VPS35, which is involved in autophagy and is implicated in PD pathogenesis [329]. The three most well studied PD-linked aSyn mutations, E46K, A30P and A53T, have been shown to promote ALP dysfunction, via either impairing autophagosome formation or inhibiting the selective removal of damaged mitochondria through mitophagy [330][331][332]. It has been previously reported that dopamine-modified aSyn inhibits CMA and this could probably shed light into the selective vulnerability of dopaminergic neurons in PD [333]. Further experiments in human iPSC-derived midbrain dopaminergic neurons revealed that disrupted hydrolase trafficking, due to aSyn overexpression, reduces lysosomal function [334]. Similarly, multiple studies suggest that there is a strong relationship between decreased β-glucocerebrosidase (GCase) activity and aSyn accumulation. In particular, heterozygote mutations in GBA1 gene encoding for β-glucocerebrosidase represent a major risk factor for PD development with a-synucleinopathy [335][336][337][338][339][340].
10. Alpha-Synuclein in the Extracellular Space
The first indication that aSyn can be secreted arose from the detection of the protein in human CSF and plasma of PD patients, indicating that aSyn can be released into the extracellular space [341][342] and can exert various deleterious effects on neighboring cells. Further studies supported that aSyn can be secreted from neuronal cells, either via vesicles or exosomes [343][344][345]. Extracellular aSyn has been the subject of intensive research in recent years, mainly due to its propensity to spread from neuron to neuron or other glial cells, as discussed in the following sections.
The major hypothesis regarding the onset and spread of aSyn pathology in a-synucleinopathies relies in the protein’s nucleation propensity that leads to the formation of aberrant aSyn species, which then spread to neighboring cells and tissues via various mechanisms. Furthermore, aSyn has been proposed to act as a “prion-like” protein since it was demonstrated that pathogenic aSyn could transfer from diseased neurons of a PD patient to the healthy transplanted ones, fourteen years after the surgical intervention [346]. Similar results were obtained by other groups in both humans and rats [347][348][349][350]. Experiments of PD and DLB patient-derived brain extracts delivered into the brain of mice and non-human primates further validated the transfer of pathological aSyn and the formation of aSyn aggregates within the recipient neurons [351][352]. Moreover, when Pre-Formed Fibrils (PFFs) were used as seeds in both in vitro and in vivo experiments, the endogenous neuronal aSyn was recruited into the formation of highly insoluble aggregates [79][353][354][355][356][357].
Various mechanisms have been proposed for aSyn spread throughout the nervous system, following its release from neurons where the protein is normally expressed. Candidate mechanisms include aSyn secretion via vesicles, exosomes or even naked protein [342][343][344][358][359][360][361] and its uptake from the cells via conventional endocytosis [362][363], passive diffusion [364], tunneling nanotubes [365], membrane penetration [173][366][367] or receptor-mediated internalization [184][368][369]. Once taken-up by recipient cells, the exogenous aSyn has been shown to trigger the endogenous aSyn accumulation via an unknown mechanism [370][371][372][373]. However, according to the prevailing hypothesis, upon the cell-internalization of aberrant aSyn conformations (oligomers or fibrils), these serve as a template for the recruitment of the endogenous monomeric aSyn into the formation of insoluble aggregates [351][353][354][355][374][375][376]. The prevalently unfolded or alpha-helical aSyn is triggered to self-assemble generating fibrils that subsequently deposit as Lewy bodies [377][378][379].
Neuron-to-neuron aSyn transmission occurs following both anterograde and retrograde axonal transport or trans-synaptic pathways [380][381][382]. Several groups have proposed that dysregulation of axonal transport is implicated in aSyn accumulation at the cell body; however, it is not clear whether PD-linked aSyn mutations play a key role in the process per se [381][383][384][385]. Notably, aSyn in its oligomeric form has been shown to interfere with microtubules and kinesin motors, thus disrupting the anterograde transport and similar results were obtained in an aSyn overexpressing mouse model for PD, as well as in patients diagnosed with the disease [386][387][388]. Additionally, it has been suggested that the variety in a-synucleinopathy phenotypes is attributed to the formation of different aSyn “strains” that display “aggressive” characteristics [389][390][391]. As a consequence of their disparate structures, these “strains” have discrete biochemical responses along the different brain regions and cell types, thus explaining the various disease manifestations of a-synucleinopathies [392][393][103][394][395][396].
References
- Iwai, A.; Masliah, E.; Yoshimoto, M.; Ge, N.; Flanagan, L.; de Silva, H.A.; Kittel, A.; Saitoh, T. The precursor protein of non-A beta component of Alzheimer’s disease amyloid is a presynaptic protein of the central nervous system. Neuron 1995, 14, 467–475.
- Withers, G.S.; George, J.M.; Banker, G.A.; Clayton, D.F. Delayed localization of synelfin (synuclein, NACP) to presynaptic terminals in cultured rat hippocampal neurons. Brain Res. Dev. Brain Res. 1997, 99, 87–94.
- Andringa, G.; Du, F.; Chase, T.N.; Bennett, M.C. Mapping of rat brain using the Synuclein-1 monoclonal antibody reveals somatodendritic expression of alpha-synuclein in populations of neurons homologous to those vulnerable to Lewy body formation in human synucleopathies. J. Neuropathol. Exp. Neurol. 2003, 62, 1060–1075.
- Barbour, R.; Kling, K.; Anderson, J.P.; Banducci, K.; Cole, T.; Diep, L.; Fox, M.; Goldstein, J.M.; Soriano, F.; Seubert, P.; et al. Red blood cells are the major source of alpha-synuclein in blood. Neurodegener. Dis. 2008, 5, 55–59.
- Askanas, V.; Engel, W.K.; Alvarez, R.B.; McFerrin, J.; Broccolini, A. Novel immunolocalization of alpha-synuclein in human muscle of inclusion-body myositis, regenerating and necrotic muscle fibers, and at neuromuscular junctions. J. Neuropathol. Exp. Neurol. 2000, 59, 592–598.
- Bottner, M.; Zorenkov, D.; Hellwig, I.; Barrenschee, M.; Harde, J.; Fricke, T.; Deuschl, G.; Egberts, J.H.; Becker, T.; Fritscher-Ravens, A.; et al. Expression pattern and localization of alpha-synuclein in the human enteric nervous system. Neurobiol. Dis. 2012, 48, 474–480.
- Aldecoa, I.; Navarro-Otano, J.; Stefanova, N.; Sprenger, F.S.; Seppi, K.; Poewe, W.; Cuatrecasas, M.; Valldeoriola, F.; Gelpi, E.; Tolosa, E. Alpha-synuclein immunoreactivity patterns in the enteric nervous system. Neurosci. Lett. 2015, 602, 145–149.
- Li, J.; Henning Jensen, P.; Dahlstrom, A. Differential localization of alpha-, beta- and gamma-synucleins in the rat CNS. Neuroscience 2002, 113, 463–478.
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Expression of alpha-synuclein is regulated in a neuronal cell type-dependent manner. Anat. Sci. Int. 2019, 94, 11–22.
- Taguchi, K.; Watanabe, Y.; Tsujimura, A.; Tanaka, M. Brain region-dependent differential expression of alpha-synuclein. J. Comp. Neurol. 2016, 524, 1236–1258.
- George, J.M.; Jin, H.; Woods, W.S.; Clayton, D.F. Characterization of a novel protein regulated during the critical period for song learning in the zebra finch. Neuron 1995, 15, 361–372.
- Maroteaux, L.; Campanelli, J.T.; Scheller, R.H. Synuclein: A neuron-specific protein localized to the nucleus and presynaptic nerve terminal. J. Neurosci. 1988, 8, 2804–2815.
- Burre, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Sudhof, T.C. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667.
- Zaltieri, M.; Grigoletto, J.; Longhena, F.; Navarria, L.; Favero, G.; Castrezzati, S.; Colivicchi, M.A.; Della Corte, L.; Rezzani, R.; Pizzi, M.; et al. alpha-synuclein and synapsin III cooperatively regulate synaptic function in dopamine neurons. J. Cell Sci. 2015, 128, 2231–2243.
- Chen, R.H.C.; Wislet-Gendebien, S.; Samuel, F.; Visanji, N.P.; Zhang, G.; Marsilio, D.; Langman, T.; Fraser, P.E.; Tandon, A. alpha-Synuclein membrane association is regulated by the Rab3a recycling machinery and presynaptic activity. J. Biol. Chem. 2013, 288, 7438–7449.
- Greten-Harrison, B.; Polydoro, M.; Morimoto-Tomita, M.; Diao, L.; Williams, A.M.; Nie, E.H.; Makani, S.; Tian, N.; Castillo, P.E.; Buchman, V.L.; et al. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2010, 107, 19573–19578.
- Burre, J.; Sharma, M.; Sudhof, T.C. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc. Natl. Acad. Sci. USA 2014, 111, E4274–E4283.
- Lautenschlager, J.; Stephens, A.D.; Fusco, G.; Strohl, F.; Curry, N.; Zacharopoulou, M.; Michel, C.H.; Laine, R.; Nespovitaya, N.; Fantham, M.; et al. C-terminal calcium binding of alpha-synuclein modulates synaptic vesicle interaction. Nat. Commun. 2018, 9, 712.
- Burre, J.; Sharma, M.; Sudhof, T.C. Definition of a molecular pathway mediating alpha-synuclein neurotoxicity. J. Neurosci. 2015, 35, 5221–5232.
- Betzer, C.; Movius, A.J.; Shi, M.; Gai, W.P.; Zhang, J.; Jensen, P.H. Identification of synaptosomal proteins binding to monomeric and oligomeric alpha-synuclein. PLoS ONE 2015, 10, e0116473.
- Scott, D.; Roy, S. alpha-Synuclein inhibits intersynaptic vesicle mobility and maintains recycling-pool homeostasis. J. Neurosci. 2012, 32, 10129–10135.
- Vargas, K.J.; Makani, S.; Davis, T.; Westphal, C.H.; Castillo, P.E.; Chandra, S.S. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J. Neurosci. 2014, 34, 9364–9376.
- Zhang, L.; Zhang, C.; Zhu, Y.; Cai, Q.; Chan, P.; Ueda, K.; Yu, S.; Yang, H. Semi-quantitative analysis of alpha-synuclein in subcellular pools of rat brain neurons: An immunogold electron microscopic study using a C-terminal specific monoclonal antibody. Brain Res. 2008, 1244, 40–52.
- Ostrerova, N.; Petrucelli, L.; Farrer, M.; Mehta, N.; Choi, P.; Hardy, J.; Wolozin, B. alpha-Synuclein shares physical and functional homology with 14-3-3 proteins. J. Neurosci. 1999, 19, 5782–5791.
- Kim, T.D.; Choi, E.; Rhim, H.; Paik, S.R.; Yang, C.H. Alpha-synuclein has structural and functional similarities to small heat shock proteins. Biochem. Biophys. Res. Commun. 2004, 324, 1352–1359.
- Chandra, S.; Gallardo, G.; Fernandez-Chacon, R.; Schluter, O.M.; Sudhof, T.C. Alpha-synuclein cooperates with CSPalpha in preventing neurodegeneration. Cell 2005, 123, 383–396.
- Kim, T.D.; Paik, S.R.; Yang, C.H. Structural and functional implications of C-terminal regions of alpha-synuclein. Biochemistry 2002, 41, 13782–13790.
- Kim, T.D.; Paik, S.R.; Yang, C.H.; Kim, J. Structural changes in alpha-synuclein affect its chaperone-like activity in vitro. Protein Sci. 2000, 9, 2489–2496.
- Park, S.M.; Jung, H.Y.; Kim, T.D.; Park, J.H.; Yang, C.H.; Kim, J. Distinct roles of the N-terminal-binding domain and the C-terminal-solubilizing domain of alpha-synuclein, a molecular chaperone. J. Biol. Chem. 2002, 277, 28512–28520.
- Rekas, A.; Ahn, K.J.; Kim, J.; Carver, J.A. The chaperone activity of alpha-synuclein: Utilizing deletion mutants to map its interaction with target proteins. Proteins 2012, 80, 1316–1325.
- Srivastava, T.; Raj, R.; Dubey, A.; Kumar, D.; Chaturvedi, R.K.; Sharma, S.K.; Priya, S. Fast kinetics of environmentally induced alpha-synuclein aggregation mediated by structural alteration in NAC region and result in structure dependent cytotoxicity. Sci. Rep. 2020, 10, 18412.
- Burre, J.; Vivona, S.; Diao, J.; Sharma, M.; Brunger, A.T.; Sudhof, T.C. Properties of native brain alpha-synuclein. Nature 2013, 498, E4–E6.
- Celej, M.S.; Sarroukh, R.; Goormaghtigh, E.; Fidelio, G.D.; Ruysschaert, J.M.; Raussens, V. Toxic prefibrillar alpha-synuclein amyloid oligomers adopt a distinctive antiparallel beta-sheet structure. Biochem. J. 2012, 443, 719–726.
- Li, H.T.; Du, H.N.; Tang, L.; Hu, J.; Hu, H.Y. Structural transformation and aggregation of human alpha-synuclein in trifluoroethanol: Non-amyloid component sequence is essential and beta-sheet formation is prerequisite to aggregation. Biopolymers 2002, 64, 221–226.
- Chaudhary, H.; Subramaniam, V.; Claessens, M. Direct visualization of model membrane remodeling by alpha-synuclein fibrillization. ChemPhysChem 2017, 18, 1620–1626.
- Galvagnion, C.; Brown, J.W.; Ouberai, M.M.; Flagmeier, P.; Vendruscolo, M.; Buell, A.K.; Sparr, E.; Dobson, C.M. Chemical properties of lipids strongly affect the kinetics of the membrane-induced aggregation of alpha-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 7065–7070.
- Galvagnion, C. The Role of Lipids Interacting with alpha-Synuclein in the Pathogenesis of Parkinson’s Disease. J. Parkinsons Dis. 2017, 7, 433–450.
- Narayanan, V.; Scarlata, S. Membrane binding and self-association of alpha-synucleins. Biochemistry 2001, 40, 9927–9934.
- Zhu, M.; Fink, A.L. Lipid binding inhibits alpha-synuclein fibril formation. J. Biol. Chem. 2003, 278, 16873–16877.
- Lee, H.J.; Choi, C.; Lee, S.J. Membrane-bound alpha-synuclein has a high aggregation propensity and the ability to seed the aggregation of the cytosolic form. J. Biol. Chem. 2002, 277, 671–678.
- Cole, N.B.; Murphy, D.D.; Grider, T.; Rueter, S.; Brasaemle, D.; Nussbaum, R.L. Lipid droplet binding and oligomerization properties of the Parkinson’s disease protein alpha-synuclein. J. Biol. Chem. 2002, 277, 6344–6352.
- Perni, M.; Galvagnion, C.; Maltsev, A.; Meisl, G.; Muller, M.B.; Challa, P.K.; Kirkegaard, J.B.; Flagmeier, P.; Cohen, S.I.; Cascella, R.; et al. A natural product inhibits the initiation of alpha-synuclein aggregation and suppresses its toxicity. Proc. Natl. Acad. Sci. USA 2017, 114, E1009–E1017.
- Fares, M.B.; Ait-Bouziad, N.; Dikiy, I.; Mbefo, M.K.; Jovicic, A.; Kiely, A.; Holton, J.L.; Lee, S.J.; Gitler, A.D.; Eliezer, D.; et al. The novel Parkinson’s disease linked mutation G51D attenuates in vitro aggregation and membrane binding of alpha-synuclein, and enhances its secretion and nuclear localization in cells. Hum. Mol. Genet. 2014, 23, 4491–4509.
- Ghosh, D.; Sahay, S.; Ranjan, P.; Salot, S.; Mohite, G.M.; Singh, P.K.; Dwivedi, S.; Carvalho, E.; Banerjee, R.; Kumar, A.; et al. The newly discovered Parkinson’s disease associated Finnish mutation (A53E) attenuates alpha-synuclein aggregation and membrane binding. Biochemistry 2014, 53, 6419–6421.
- Jensen, P.H.; Nielsen, M.S.; Jakes, R.; Dotti, C.G.; Goedert, M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson’s disease mutation. J. Biol. Chem. 1998, 273, 26292–26294.
- Jo, E.; Fuller, N.; Rand, R.P.; St George-Hyslop, P.; Fraser, P.E. Defective membrane interactions of familial Parkinson’s disease mutant A30P alpha-synuclein. J. Mol. Biol. 2002, 315, 799–807.
- Tsigelny, I.F.; Sharikov, Y.; Kouznetsova, V.L.; Greenberg, J.P.; Wrasidlo, W.; Overk, C.; Gonzalez, T.; Trejo, M.; Spencer, B.; Kosberg, K.; et al. Molecular determinants of alpha-synuclein mutants’ oligomerization and membrane interactions. ACS Chem. Neurosci. 2015, 6, 403–416.
- Eliezer, D.; Kutluay, E.; Bussell, R., Jr.; Browne, G. Conformational properties of alpha-synuclein in its free and lipid-associated states. J. Mol. Biol. 2001, 307, 1061–1073.
- Weinreb, P.H.; Zhen, W.; Poon, A.W.; Conway, K.A.; Lansbury, P.T., Jr. NACP, a protein implicated in Alzheimer’s disease and learning, is natively unfolded. Biochemistry 1996, 35, 13709–13715.
- Killinger, B.A.; Melki, R.; Brundin, P.; Kordower, J.H. Endogenous alpha-synuclein monomers, oligomers and resulting pathology: Let’s talk about the lipids in the room. NPJ Parkinsons Dis. 2019, 5, 23.
- Fauvet, B.; Mbefo, M.K.; Fares, M.B.; Desobry, C.; Michael, S.; Ardah, M.T.; Tsika, E.; Coune, P.; Prudent, M.; Lion, N.; et al. alpha-Synuclein in central nervous system and from erythrocytes, mammalian cells, and Escherichia coli exists predominantly as disordered monomer. J. Biol. Chem. 2012, 287, 15345–15364.
- Bartels, T.; Choi, J.G.; Selkoe, D.J. alpha-Synuclein occurs physiologically as a helically folded tetramer that resists aggregation. Nature 2011, 477, 107–110.
- Wang, W.; Perovic, I.; Chittuluru, J.; Kaganovich, A.; Nguyen, L.T.; Liao, J.; Auclair, J.R.; Johnson, D.; Landeru, A.; Simorellis, A.K.; et al. A soluble alpha-synuclein construct forms a dynamic tetramer. Proc. Natl. Acad. Sci. USA 2011, 108, 17797–17802.
- Dettmer, U.; Newman, A.J.; Luth, E.S.; Bartels, T.; Selkoe, D. In vivo cross-linking reveals principally oligomeric forms of alpha-synuclein and beta-synuclein in neurons and non-neural cells. J. Biol. Chem. 2013, 288, 6371–6385.
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449.
- Jo, E.; McLaurin, J.; Yip, C.M.; St George-Hyslop, P.; Fraser, P.E. Alpha-Synuclein membrane interactions and lipid specificity. J. Biol. Chem. 2000, 275, 34328–34334.
- Trexler, A.J.; Rhoades, E. Alpha-synuclein binds large unilamellar vesicles as an extended helix. Biochemistry 2009, 48, 2304–2306.
- Jao, C.C.; Der-Sarkissian, A.; Chen, J.; Langen, R. Structure of membrane-bound alpha-synuclein studied by site-directed spin labeling. Proc. Natl. Acad. Sci. USA 2004, 101, 8331–8336.
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Sudhof, T.C. A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem. 2003, 278, 15313–15318.
- Drescher, M.; Veldhuis, G.; van Rooijen, B.D.; Milikisyants, S.; Subramaniam, V.; Huber, M. Antiparallel arrangement of the helices of vesicle-bound alpha-synuclein. J. Am. Chem. Soc. 2008, 130, 7796–7797.
- Sharon, R.; Goldberg, M.S.; Bar-Josef, I.; Betensky, R.A.; Shen, J.; Selkoe, D.J. alpha-Synuclein occurs in lipid-rich high molecular weight complexes, binds fatty acids, and shows homology to the fatty acid-binding proteins. Proc. Natl. Acad. Sci. USA 2001, 98, 9110–9115.
- Lucke, C.; Gantz, D.L.; Klimtchuk, E.; Hamilton, J.A. Interactions between fatty acids and alpha-synuclein. J. Lipid Res. 2006, 47, 1714–1724.
- Madine, J.; Doig, A.J.; Middleton, D.A. A study of the regional effects of alpha-synuclein on the organization and stability of phospholipid bilayers. Biochemistry 2006, 45, 5783–5792.
- Adamczyk, A.; Kacprzak, M.; Kazmierczak, A. Alpha-synuclein decreases arachidonic acid incorporation into rat striatal synaptoneurosomes. Folia Neuropathol. 2007, 45, 230–235.
- Castagnet, P.I.; Golovko, M.Y.; Barcelo-Coblijn, G.C.; Nussbaum, R.L.; Murphy, E.J. Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J. Neurochem. 2005, 94, 839–849.
- Barcelo-Coblijn, G.; Golovko, M.Y.; Weinhofer, I.; Berger, J.; Murphy, E.J. Brain neutral lipids mass is increased in alpha-synuclein gene-ablated mice. J. Neurochem. 2007, 101, 132–141.
- Alza, N.P.; Conde, M.A.; Scodelaro-Bilbao, P.G.; Salvador, G.A. Neutral lipids as early biomarkers of cellular fate: The case of alpha-synuclein overexpression. Cell Death Dis. 2021, 12, 52.
- Ruf, V.C.; Nubling, G.S.; Willikens, S.; Shi, S.; Schmidt, F.; Levin, J.; Botzel, K.; Kamp, F.; Giese, A. Different effects of alpha-synuclein mutants on lipid binding and aggregation detected by single molecule fluorescence spectroscopy and ThT fluorescence-based measurements. ACS Chem. Neurosci. 2019, 10, 1649–1659.
- Conde, M.A.; Alza, N.P.; Iglesias Gonzalez, P.A.; Scodelaro Bilbao, P.G.; Sanchez Campos, S.; Uranga, R.M.; Salvador, G.A. Phospholipase D1 downregulation by alpha-synuclein: Implications for neurodegeneration in Parkinson’s disease. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 639–650.
- Ahn, B.H.; Rhim, H.; Kim, S.Y.; Sung, Y.M.; Lee, M.Y.; Choi, J.Y.; Wolozin, B.; Chang, J.S.; Lee, Y.H.; Kwon, T.K.; et al. alpha-Synuclein interacts with phospholipase D isozymes and inhibits pervanadate-induced phospholipase D activation in human embryonic kidney-293 cells. J. Biol. Chem. 2002, 277, 12334–12342.
- Jenco, J.M.; Rawlingson, A.; Daniels, B.; Morris, A.J. Regulation of phospholipase D2: Selective inhibition of mammalian phospholipase D isoenzymes by alpha- and beta-synucleins. Biochemistry 1998, 37, 4901–4909.
- Payton, J.E.; Perrin, R.J.; Woods, W.S.; George, J.M. Structural determinants of PLD2 inhibition by alpha-synuclein. J. Mol. Biol. 2004, 337, 1001–1009.
- Rappley, I.; Gitler, A.D.; Selvy, P.E.; LaVoie, M.J.; Levy, B.D.; Brown, H.A.; Lindquist, S.; Selkoe, D.J. Evidence that alpha-synuclein does not inhibit phospholipase D. Biochemistry 2009, 48, 1077–1083.
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603.
- Sode, K.; Ochiai, S.; Kobayashi, N.; Usuzaka, E. Effect of reparation of repeat sequences in the human alpha-synuclein on fibrillation ability. Int. J. Biol. Sci. 2006, 3, 1–7.
- Giasson, B.I.; Murray, I.V.; Trojanowski, J.Q.; Lee, V.M. A hydrophobic stretch of 12 amino acid residues in the middle of alpha-synuclein is essential for filament assembly. J. Biol. Chem. 2001, 276, 2380–2386.
- Rodriguez, J.A.; Ivanova, M.I.; Sawaya, M.R.; Cascio, D.; Reyes, F.E.; Shi, D.; Sangwan, S.; Guenther, E.L.; Johnson, L.M.; Zhang, M.; et al. Structure of the toxic core of alpha-synuclein from invisible crystals. Nature 2015, 525, 486–490.
- Longhena, F.; Faustini, G.; Missale, C.; Pizzi, M.; Spano, P.; Bellucci, A. The Contribution of alpha-Synuclein Spreading to Parkinson’s Disease Synaptopathy. Neural. Plast. 2017, 2017, 5012129.
- Volpicelli-Daley, L.A.; Luk, K.C.; Patel, T.P.; Tanik, S.A.; Riddle, D.M.; Stieber, A.; Meaney, D.F.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils induce Lewy body pathology leading to synaptic dysfunction and neuron death. Neuron 2011, 72, 57–71.
- Appel-Cresswell, S.; Vilarino-Guell, C.; Encarnacion, M.; Sherman, H.; Yu, I.; Shah, B.; Weir, D.; Thompson, C.; Szu-Tu, C.; Trinh, J.; et al. Alpha-synuclein p.H50Q, a novel pathogenic mutation for Parkinson’s disease. Mov. Disord. 2013, 28, 811–813.
- Kiely, A.P.; Asi, Y.T.; Kara, E.; Limousin, P.; Ling, H.; Lewis, P.; Proukakis, C.; Quinn, N.; Lees, A.J.; Hardy, J.; et al. alpha-Synucleinopathy associated with G51D SNCA mutation: A link between Parkinson’s disease and multiple system atrophy? Acta Neuropathol. 2013, 125, 753–769.
- Hoffman-Zacharska, D.; Koziorowski, D.; Ross, O.A.; Milewski, M.; Poznanski, J.A.; Jurek, M.; Wszolek, Z.K.; Soto-Ortolaza, A.; Awek, J.A.S.; Janik, P.; et al. Novel A18T and pA29S substitutions in alpha-synuclein may be associated with sporadic Parkinson’s disease. Parkinsonism Relat. Disord. 2013, 19, 1057–1060.
- Conway, K.A.; Lee, S.J.; Rochet, J.C.; Ding, T.T.; Williamson, R.E.; Lansbury, P.T., Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc. Natl. Acad. Sci. USA 2000, 97, 571–576.
- Conway, K.A.; Harper, J.D.; Lansbury, P.T. Accelerated in vitro fibril formation by a mutant alpha-synuclein linked to early-onset Parkinson disease. Nat. Med. 1998, 4, 1318–1320.
- Bussell, R., Jr.; Eliezer, D. Residual structure and dynamics in Parkinson’s disease-associated mutants of alpha-synuclein. J. Biol. Chem. 2001, 276, 45996–46003.
- Lazaro, D.F.; Rodrigues, E.F.; Langohr, R.; Shahpasandzadeh, H.; Ribeiro, T.; Guerreiro, P.; Gerhardt, E.; Krohnert, K.; Klucken, J.; Pereira, M.D.; et al. Systematic comparison of the effects of alpha-synuclein mutations on its oligomerization and aggregation. PLoS Genet. 2014, 10, e1004741.
- Fredenburg, R.A.; Rospigliosi, C.; Meray, R.K.; Kessler, J.C.; Lashuel, H.A.; Eliezer, D.; Lansbury, P.T., Jr. The impact of the E46K mutation on the properties of alpha-synuclein in its monomeric and oligomeric states. Biochemistry 2007, 46, 7107–7118.
- Rospigliosi, C.C.; McClendon, S.; Schmid, A.W.; Ramlall, T.F.; Barre, P.; Lashuel, H.A.; Eliezer, D. E46K Parkinson’s-linked mutation enhances C-terminal-to-N-terminal contacts in alpha-synuclein. J. Mol. Biol. 2009, 388, 1022–1032.
- Khalaf, O.; Fauvet, B.; Oueslati, A.; Dikiy, I.; Mahul-Mellier, A.L.; Ruggeri, F.S.; Mbefo, M.K.; Vercruysse, F.; Dietler, G.; Lee, S.J.; et al. The H50Q mutation enhances alpha-synuclein aggregation, secretion, and toxicity. J. Biol. Chem. 2014, 289, 21856–21876.
- Sanjeev, A.; Mattaparthi, V.S.K. Computational investigation on the effects of H50Q and G51D mutations on the alpha-Synuclein aggregation propensity. J. Biomol. Struct. Dyn. 2018, 36, 2224–2236.
- Lesage, S.; Anheim, M.; Letournel, F.; Bousset, L.; Honore, A.; Rozas, N.; Pieri, L.; Madiona, K.; Durr, A.; Melki, R.; et al. G51D alpha-synuclein mutation causes a novel parkinsonian-pyramidal syndrome. Ann. Neurol. 2013, 73, 459–471.
- Rutherford, N.J.; Moore, B.D.; Golde, T.E.; Giasson, B.I. Divergent effects of the H50Q and G51D SNCA mutations on the aggregation of alpha-synuclein. J. Neurochem. 2014, 131, 859–867.
- Rutherford, N.J.; Giasson, B.I. The A53E alpha-synuclein pathological mutation demonstrates reduced aggregation propensity in vitro and in cell culture. Neurosci. Lett. 2015, 597, 43–48.
- Pasanen, P.; Myllykangas, L.; Siitonen, M.; Raunio, A.; Kaakkola, S.; Lyytinen, J.; Tienari, P.J.; Poyhonen, M.; Paetau, A. Novel alpha-synuclein mutation A53E associated with atypical multiple system atrophy and Parkinson’s disease-type pathology. Neurobiol. Aging 2014, 35, 2180.e1.
- Ozawa, T.; Takano, H.; Onodera, O.; Kobayashi, H.; Ikeuchi, T.; Koide, R.; Okuizumi, K.; Shimohata, T.; Wakabayashi, K.; Takahashi, H.; et al. No mutation in the entire coding region of the alpha-synuclein gene in pathologically confirmed cases of multiple system atrophy. Neurosci. Lett. 1999, 270, 110–112.
- Narhi, L.; Wood, S.J.; Steavenson, S.; Jiang, Y.; Wu, G.M.; Anafi, D.; Kaufman, S.A.; Martin, F.; Sitney, K.; Denis, P.; et al. Both familial Parkinson’s disease mutations accelerate alpha-synuclein aggregation. J. Biol. Chem. 1999, 274, 9843–9846.
- Pandey, N.; Schmidt, R.E.; Galvin, J.E. The alpha-synuclein mutation E46K promotes aggregation in cultured cells. Exp. Neurol. 2006, 197, 515–520.
- Smith, W.W.; Margolis, R.L.; Li, X.; Troncoso, J.C.; Lee, M.K.; Dawson, V.L.; Dawson, T.M.; Iwatsubo, T.; Ross, C.A. Alpha-synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH-SY5Y cells. J. Neurosci. 2005, 25, 5544–5552.
- Sugeno, N.; Takeda, A.; Hasegawa, T.; Kobayashi, M.; Kikuchi, A.; Mori, F.; Wakabayashi, K.; Itoyama, Y. Serine 129 phosphorylation of alpha-synuclein induces unfolded protein response-mediated cell death. J. Biol. Chem. 2008, 283, 23179–23188.
- Ma, M.R.; Hu, Z.W.; Zhao, Y.F.; Chen, Y.X.; Li, Y.M. Phosphorylation induces distinct alpha-synuclein strain formation. Sci. Rep. 2016, 6, 37130.
- Sato, H.; Arawaka, S.; Hara, S.; Fukushima, S.; Koga, K.; Koyama, S.; Kato, T. Authentically phosphorylated alpha-synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson’s disease. J. Neurosci. 2011, 31, 16884–16894.
- Chen, L.; Periquet, M.; Wang, X.; Negro, A.; McLean, P.J.; Hyman, B.T.; Feany, M.B. Tyrosine and serine phosphorylation of alpha-synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J. Clin. Investig. 2009, 119, 3257–3265.
- Azeredo da Silveira, S.; Schneider, B.L.; Cifuentes-Diaz, C.; Sage, D.; Abbas-Terki, T.; Iwatsubo, T.; Unser, M.; Aebischer, P. Phosphorylation does not prompt, nor prevent, the formation of alpha-synuclein toxic species in a rat model of Parkinson’s disease. Hum. Mol. Genet. 2009, 18, 872–887.
- Mbefo, M.K.; Paleologou, K.E.; Boucharaba, A.; Oueslati, A.; Schell, H.; Fournier, M.; Olschewski, D.; Yin, G.; Zweckstetter, M.; Masliah, E.; et al. Phosphorylation of synucleins by members of the Polo-like kinase family. J. Biol. Chem. 2010, 285, 2807–2822.
- Chen, L.; Feany, M.B. Alpha-synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat. Neurosci. 2005, 8, 657–663.
- Paleologou, K.E.; Schmid, A.W.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fredenburg, R.A.; Lansbury, P.T., Jr.; Fernandez, C.O.; Eliezer, D.; Zweckstetter, M.; et al. Phosphorylation at Ser-129 but not the phosphomimics S129E/D inhibits the fibrillation of alpha-synuclein. J. Biol. Chem. 2008, 283, 16895–16905.
- Wu, B.; Liu, Q.; Duan, C.; Li, Y.; Yu, S.; Chan, P.; Ueda, K.; Yang, H. Phosphorylation of alpha-synuclein upregulates tyrosine hydroxylase activity in MN9D cells. Acta Histochem. 2011, 113, 32–35.
- Kuwahara, T.; Tonegawa, R.; Ito, G.; Mitani, S.; Iwatsubo, T. Phosphorylation of alpha-synuclein protein at Ser-129 reduces neuronal dysfunction by lowering its membrane binding property in Caenorhabditis elegans. J. Biol. Chem. 2012, 287, 7098–7109.
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; de Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M.; et al. Phosphorylation of Ser-129 is the dominant pathological modification of alpha-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752.
- Fujiwara, H.; Hasegawa, M.; Dohmae, N.; Kawashima, A.; Masliah, E.; Goldberg, M.S.; Shen, J.; Takio, K.; Iwatsubo, T. alpha-Synuclein is phosphorylated in synucleinopathy lesions. Nat. Cell Biol. 2002, 4, 160–164.
- Sato, H.; Kato, T.; Arawaka, S. The role of Ser129 phosphorylation of alpha-synuclein in neurodegeneration of Parkinson’s disease: A review of in vivo models. Rev. Neurosci. 2013, 24, 115–123.
- Walker, D.G.; Lue, L.F.; Adler, C.H.; Shill, H.A.; Caviness, J.N.; Sabbagh, M.N.; Akiyama, H.; Serrano, G.E.; Sue, L.I.; Beach, T.G. Changes in properties of serine 129 phosphorylated alpha-synuclein with progression of Lewy-type histopathology in human brains. Exp. Neurol. 2013, 240, 190–204.
- Paleologou, K.E.; Oueslati, A.; Shakked, G.; Rospigliosi, C.C.; Kim, H.Y.; Lamberto, G.R.; Fernandez, C.O.; Schmid, A.; Chegini, F.; Gai, W.P.; et al. Phosphorylation at S87 is enhanced in synucleinopathies, inhibits alpha-synuclein oligomerization, and influences synuclein-membrane interactions. J. Neurosci. 2010, 30, 3184–3198.
- Xu, Y.; Deng, Y.; Qing, H. The phosphorylation of alpha-synuclein: Development and implication for the mechanism and therapy of the Parkinson’s disease. J. Neurochem. 2015, 135, 4–18.
- Lu, Y.; Prudent, M.; Fauvet, B.; Lashuel, H.A.; Girault, H.H. Phosphorylation of alpha-Synuclein at Y125 and S129 alters its metal binding properties: Implications for understanding the role of alpha-Synuclein in the pathogenesis of Parkinson’s Disease and related disorders. ACS Chem. Neurosci. 2011, 2, 667–675.
- Fayyad, M.; Erskine, D.; Majbour, N.K.; Vaikath, N.N.; Ghanem, S.S.; Sudhakaran, I.P.; Abdesselem, H.; Lamprokostopoulou, A.; Vekrellis, K.; Morris, C.M.; et al. Investigating the presence of doubly phosphorylated alpha-synuclein at tyrosine 125 and serine 129 in idiopathic Lewy body diseases. Brain Pathol. 2020, 30, 831–843.
- Giasson, B.I.; Duda, J.E.; Murray, I.V.; Chen, Q.; Souza, J.M.; Hurtig, H.I.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science 2000, 290, 985–989.
- Danielson, S.R.; Held, J.M.; Schilling, B.; Oo, M.; Gibson, B.W.; Andersen, J.K. Preferentially increased nitration of alpha-synuclein at tyrosine-39 in a cellular oxidative model of Parkinson’s disease. Anal. Chem. 2009, 81, 7823–7828.
- McCormack, A.L.; Mak, S.K.; Di Monte, D.A. Increased alpha-synuclein phosphorylation and nitration in the aging primate substantia nigra. Cell Death Dis. 2012, 3, e315.
- Yu, Z.; Xu, X.; Xiang, Z.; Zhou, J.; Zhang, Z.; Hu, C.; He, C. Nitrated alpha-synuclein induces the loss of dopaminergic neurons in the substantia nigra of rats. PLoS ONE 2010, 5, e9956.
- Souza, J.M.; Peluffo, G.; Radi, R. Protein tyrosine nitration—Functional alteration or just a biomarker? Free Radic. Biol. Med. 2008, 45, 357–366.
- Hodara, R.; Norris, E.H.; Giasson, B.I.; Mishizen-Eberz, A.J.; Lynch, D.R.; Lee, V.M.; Ischiropoulos, H. Functional consequences of alpha-synuclein tyrosine nitration: Diminished binding to lipid vesicles and increased fibril formation. J. Biol. Chem. 2004, 279, 47746–47753.
- Takahashi, T.; Yamashita, H.; Nakamura, T.; Nagano, Y.; Nakamura, S. Tyrosine 125 of alpha-synuclein plays a critical role for dimerization following nitrative stress. Brain Res. 2002, 938, 73–80.
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable alpha -synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349.
- Fernandez, E.; Garcia-Moreno, J.M.; Martin de Pablos, A.; Chacon, J. May the evaluation of nitrosative stress through selective increase of 3-nitrotyrosine proteins other than nitroalbumin and dominant tyrosine-125/136 nitrosylation of serum alpha-synuclein serve for diagnosis of sporadic Parkinson’s disease? Antioxid. Redox. Signal. 2013, 19, 912–918.
- Grice, G.L.; Nathan, J.A. The recognition of ubiquitinated proteins by the proteasome. Cell Mol. Life Sci. 2016, 73, 3497–3506.
- Pickart, C.M.; Eddins, M.J. Ubiquitin: Structures, functions, mechanisms. Biochim. Biophys. Acta 2004, 1695, 55–72.
- Tofaris, G.K.; Razzaq, A.; Ghetti, B.; Lilley, K.S.; Spillantini, M.G. Ubiquitination of alpha-synuclein in Lewy bodies is a pathological event not associated with impairment of proteasome function. J. Biol. Chem. 2003, 278, 44405–44411.
- Hasegawa, M.; Fujiwara, H.; Nonaka, T.; Wakabayashi, K.; Takahashi, H.; Lee, V.M.; Trojanowski, J.Q.; Mann, D.; Iwatsubo, T. Phosphorylated alpha-synuclein is ubiquitinated in alpha-synucleinopathy lesions. J. Biol. Chem. 2002, 277, 49071–49076.
- Liani, E.; Eyal, A.; Avraham, E.; Shemer, R.; Szargel, R.; Berg, D.; Bornemann, A.; Riess, O.; Ross, C.A.; Rott, R.; et al. Ubiquitylation of synphilin-1 and alpha-synuclein by SIAH and its presence in cellular inclusions and Lewy bodies imply a role in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 5500–5505.
- Shin, Y.; Klucken, J.; Patterson, C.; Hyman, B.T.; McLean, P.J. The co-chaperone carboxyl terminus of Hsp70-interacting protein (CHIP) mediates alpha-synuclein degradation decisions between proteasomal and lysosomal pathways. J. Biol. Chem. 2005, 280, 23727–23734.
- Tofaris, G.K.; Kim, H.T.; Hourez, R.; Jung, J.W.; Kim, K.P.; Goldberg, A.L. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc. Natl. Acad. Sci. USA 2011, 108, 17004–17009.
- Lee, J.T.; Wheeler, T.C.; Li, L.; Chin, L.S. Ubiquitination of alpha-synuclein by Siah-1 promotes alpha-synuclein aggregation and apoptotic cell death. Hum. Mol. Genet. 2008, 17, 906–917.
- Rott, R.; Szargel, R.; Haskin, J.; Shani, V.; Shainskaya, A.; Manov, I.; Liani, E.; Avraham, E.; Engelender, S. Monoubiquitylation of alpha-synuclein by seven in absentia homolog (SIAH) promotes its aggregation in dopaminergic cells. J. Biol. Chem. 2008, 283, 3316–3328.
- Kalia, L.V.; Kalia, S.K.; Chau, H.; Lozano, A.M.; Hyman, B.T.; McLean, P.J. Ubiquitinylation of alpha-synuclein by carboxyl terminus Hsp70-interacting protein (CHIP) is regulated by Bcl-2-associated athanogene 5 (BAG5). PLoS ONE 2011, 6, e14695.
- Meier, F.; Abeywardana, T.; Dhall, A.; Marotta, N.P.; Varkey, J.; Langen, R.; Chatterjee, C.; Pratt, M.R. Semisynthetic, site-specific ubiquitin modification of alpha-synuclein reveals differential effects on aggregation. J. Am. Chem. Soc. 2012, 134, 5468–5471.
- Weetman, J.; Wong, M.B.; Sharry, S.; Rcom-H’cheo-Gauthier, A.; Gai, W.P.; Meedeniya, A.; Pountney, D.L. Increased SUMO-1 expression in the unilateral rotenone-lesioned mouse model of Parkinson’s disease. Neurosci. Lett. 2013, 544, 119–124.
- Wong, M.B.; Goodwin, J.; Norazit, A.; Meedeniya, A.C.; Richter-Landsberg, C.; Gai, W.P.; Pountney, D.L. SUMO-1 is associated with a subset of lysosomes in glial protein aggregate diseases. Neurotox. Res. 2013, 23, 1–21.
- Pountney, D.L.; Chegini, F.; Shen, X.; Blumbergs, P.C.; Gai, W.P. SUMO-1 marks subdomains within glial cytoplasmic inclusions of multiple system atrophy. Neurosci. Lett. 2005, 381, 74–79.
- Rott, R.; Szargel, R.; Shani, V.; Hamza, H.; Savyon, M.; Abd Elghani, F.; Bandopadhyay, R.; Engelender, S. SUMOylation and ubiquitination reciprocally regulate alpha-synuclein degradation and pathological aggregation. Proc. Natl. Acad. Sci. USA 2017, 114, 13176–13181.
- Abeywardana, T.; Pratt, M.R. Extent of inhibition of alpha-synuclein aggregation in vitro by SUMOylation is conjugation site- and SUMO isoform-selective. Biochemistry 2015, 54, 959–961.
- Krumova, P.; Meulmeester, E.; Garrido, M.; Tirard, M.; Hsiao, H.H.; Bossis, G.; Urlaub, H.; Zweckstetter, M.; Kugler, S.; Melchior, F.; et al. Sumoylation inhibits alpha-synuclein aggregation and toxicity. J. Cell Biol. 2011, 194, 49–60.
- Vinueza-Gavilanes, R.; Inigo-Marco, I.; Larrea, L.; Lasa, M.; Carte, B.; Santamaria, E.; Fernandez-Irigoyen, J.; Bugallo, R.; Aragon, T.; Aldabe, R.; et al. N-terminal acetylation mutants affect alpha-synuclein stability, protein levels and neuronal toxicity. Neurobiol. Dis. 2020, 137, 104781.
- Iyer, A.; Roeters, S.J.; Schilderink, N.; Hommersom, B.; Heeren, R.M.; Woutersen, S.; Claessens, M.M.; Subramaniam, V. The impact of N-terminal acetylation of alpha-synuclein on phospholipid membrane binding and fibril structure. J. Biol. Chem. 2016, 291, 21110–21122.
- Kang, L.; Moriarty, G.M.; Woods, L.A.; Ashcroft, A.E.; Radford, S.E.; Baum, J. N-terminal acetylation of alpha-synuclein induces increased transient helical propensity and decreased aggregation rates in the intrinsically disordered monomer. Protein Sci. 2012, 21, 911–917.
- Runfola, M.; De Simone, A.; Vendruscolo, M.; Dobson, C.M.; Fusco, G. The N-terminal acetylation of alpha-synuclein changes the affinity for lipid membranes but not the structural properties of the bound state. Sci. Rep. 2020, 10, 204.
- Bu, B.; Tong, X.; Li, D.; Hu, Y.; He, W.; Zhao, C.; Hu, R.; Li, X.; Shao, Y.; Liu, C.; et al. N-terminal acetylation preserves alpha-synuclein from oligomerization by blocking intermolecular hydrogen bonds. ACS Chem. Neurosci. 2017, 8, 2145–2151.
- Bartels, T.; Kim, N.C.; Luth, E.S.; Selkoe, D.J. N-alpha-acetylation of alpha-synuclein increases its helical folding propensity, GM1 binding specificity and resistance to aggregation. PLoS ONE 2014, 9, e103727.
- Wang, S.; Yang, F.; Petyuk, V.A.; Shukla, A.K.; Monroe, M.E.; Gritsenko, M.A.; Rodland, K.D.; Smith, R.D.; Qian, W.J.; Gong, C.X.; et al. Quantitative proteomics identifies altered O-GlcNAcylation of structural, synaptic and memory-associated proteins in Alzheimer’s disease. J. Pathol. 2017, 243, 78–88.
- Alfaro, J.F.; Gong, C.X.; Monroe, M.E.; Aldrich, J.T.; Clauss, T.R.; Purvine, S.O.; Wang, Z.; Camp, D.G., 2nd; Shabanowitz, J.; Stanley, P.; et al. Tandem mass spectrometry identifies many mouse brain O-GlcNAcylated proteins including EGF domain-specific O-GlcNAc transferase targets. Proc. Natl. Acad. Sci. USA 2012, 109, 7280–7285.
- Morris, M.; Knudsen, G.M.; Maeda, S.; Trinidad, J.C.; Ioanoviciu, A.; Burlingame, A.L.; Mucke, L. Tau post-translational modifications in wild-type and human amyloid precursor protein transgenic mice. Nat. Neurosci. 2015, 18, 1183–1189.
- Wang, Z.; Park, K.; Comer, F.; Hsieh-Wilson, L.C.; Saudek, C.D.; Hart, G.W. Site-specific GlcNAcylation of human erythrocyte proteins: Potential biomarker(s) for diabetes. Diabetes 2009, 58, 309–317.
- Wang, Z.; Udeshi, N.D.; O’Malley, M.; Shabanowitz, J.; Hunt, D.F.; Hart, G.W. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol. Cell Proteom. 2010, 9, 153–160.
- Zhang, J.; Lei, H.; Chen, Y.; Ma, Y.T.; Jiang, F.; Tan, J.; Zhang, Y.; Li, J.D. Enzymatic O-GlcNAcylation of alpha-synuclein reduces aggregation and increases SDS-resistant soluble oligomers. Neurosci. Lett. 2017, 655, 90–94.
- Levine, P.M.; Galesic, A.; Balana, A.T.; Mahul-Mellier, A.L.; Navarro, M.X.; De Leon, C.A.; Lashuel, H.A.; Pratt, M.R. alpha-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 1511–1519.
- Lewis, Y.E.; Galesic, A.; Levine, P.M.; De Leon, C.A.; Lamiri, N.; Brennan, C.K.; Pratt, M.R. O-GlcNAcylation of alpha-synuclein at serine 87 reduces aggregation without affecting membrane binding. ACS Chem. Biol. 2017, 12, 1020–1027.
- Marotta, N.P.; Lin, Y.H.; Lewis, Y.E.; Ambroso, M.R.; Zaro, B.W.; Roth, M.T.; Arnold, D.B.; Langen, R.; Pratt, M.R. O-GlcNAc modification blocks the aggregation and toxicity of the protein alpha-synuclein associated with Parkinson’s disease. Nat. Chem. 2015, 7, 913–920.
- Li, W.; West, N.; Colla, E.; Pletnikova, O.; Troncoso, J.C.; Marsh, L.; Dawson, T.M.; Jakala, P.; Hartmann, T.; Price, D.L.; et al. Aggregation promoting C-terminal truncation of alpha-synuclein is a normal cellular process and is enhanced by the familial Parkinson’s disease-linked mutations. Proc. Natl. Acad. Sci. USA 2005, 102, 2162–2167.
- Kellie, J.F.; Higgs, R.E.; Ryder, J.W.; Major, A.; Beach, T.G.; Adler, C.H.; Merchant, K.; Knierman, M.D. Quantitative measurement of intact alpha-synuclein proteoforms from post-mortem control and Parkinson’s disease brain tissue by intact protein mass spectrometry. Sci. Rep. 2014, 4, 5797.
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884.
- Ulusoy, A.; Febbraro, F.; Jensen, P.H.; Kirik, D.; Romero-Ramos, M. Co-expression of C-terminal truncated alpha-synuclein enhances full-length alpha-synuclein-induced pathology. Eur. J. Neurosci. 2010, 32, 409–422.
- Hall, K.; Yang, S.; Sauchanka, O.; Spillantini, M.G.; Anichtchik, O. Behavioural deficits in transgenic mice expressing human truncated (1-120 amino acid) alpha-synuclein. Exp. Neurol. 2015, 264, 8–13.
- Wang, W.; Nguyen, L.T.; Burlak, C.; Chegini, F.; Guo, F.; Chataway, T.; Ju, S.; Fisher, O.S.; Miller, D.W.; Datta, D.; et al. Caspase-1 causes truncation and aggregation of the Parkinson’s disease-associated protein alpha-synuclein. Proc. Natl. Acad. Sci. USA 2016, 113, 9587–9592.
- Iyer, A.; Roeters, S.J.; Kogan, V.; Woutersen, S.; Claessens, M.; Subramaniam, V. C-terminal truncated alpha-synuclein fibrils contain strongly twisted beta-sheets. J. Am. Chem. Soc. 2017, 139, 15392–15400.
- Ma, L.; Yang, C.; Zhang, X.; Li, Y.; Wang, S.; Zheng, L.; Huang, K. C-terminal truncation exacerbates the aggregation and cytotoxicity of alpha-Synuclein: A vicious cycle in Parkinson’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 3714–3725.
- Terada, M.; Suzuki, G.; Nonaka, T.; Kametani, F.; Tamaoka, A.; Hasegawa, M. The effect of truncation on prion-like properties of alpha-synuclein. J. Biol. Chem. 2018, 293, 13910–13920.
- Tofaris, G.K.; Garcia Reitbock, P.; Humby, T.; Lambourne, S.L.; O’Connell, M.; Ghetti, B.; Gossage, H.; Emson, P.C.; Wilkinson, L.S.; Goedert, M.; et al. Pathological changes in dopaminergic nerve cells of the substantia nigra and olfactory bulb in mice transgenic for truncated human alpha-synuclein(1-120): Implications for Lewy body disorders. J. Neurosci. 2006, 26, 3942–3950.
- Hoyer, W.; Cherny, D.; Subramaniam, V.; Jovin, T.M. Impact of the acidic C-terminal region comprising amino acids 109-140 on alpha-synuclein aggregation in vitro. Biochemistry 2004, 43, 16233–16242.
- Murray, I.V.; Giasson, B.I.; Quinn, S.M.; Koppaka, V.; Axelsen, P.H.; Ischiropoulos, H.; Trojanowski, J.Q.; Lee, V.M. Role of alpha-synuclein carboxy-terminus on fibril formation in vitro. Biochemistry 2003, 42, 8530–8540.
- Tsigelny, I.F.; Sharikov, Y.; Wrasidlo, W.; Gonzalez, T.; Desplats, P.A.; Crews, L.; Spencer, B.; Masliah, E. Role of alpha-synuclein penetration into the membrane in the mechanisms of oligomer pore formation. FEBS J. 2012, 279, 1000–1013.
- Fantini, J.; Yahi, N. The driving force of alpha-synuclein insertion and amyloid channel formation in the plasma membrane of neural cells: Key role of ganglioside- and cholesterol-binding domains. Adv. Exp. Med. Biol. 2013, 991, 15–26.
- Zakharov, S.D.; Hulleman, J.D.; Dutseva, E.A.; Antonenko, Y.N.; Rochet, J.C.; Cramer, W.A. Helical alpha-synuclein forms highly conductive ion channels. Biochemistry 2007, 46, 14369–14379.
- Quist, A.; Doudevski, I.; Lin, H.; Azimova, R.; Ng, D.; Frangione, B.; Kagan, B.; Ghiso, J.; Lal, R. Amyloid ion channels: A common structural link for protein-misfolding disease. Proc. Natl. Acad. Sci. USA 2005, 102, 10427–10432.
- Volles, M.J.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein is sensitive to Parkinson’s disease-linked mutations and occurs by a pore-like mechanism. Biochemistry 2002, 41, 4595–4602.
- Stockl, M.T.; Zijlstra, N.; Subramaniam, V. alpha-Synuclein oligomers: An amyloid pore? Insights into mechanisms of alpha-synuclein oligomer-lipid interactions. Mol. Neurobiol. 2013, 47, 613–621.
- Di Pasquale, E.; Fantini, J.; Chahinian, H.; Maresca, M.; Taieb, N.; Yahi, N. Altered ion channel formation by the Parkinson’s-disease-linked E46K mutant of alpha-synuclein is corrected by GM3 but not by GM1 gangliosides. J. Mol. Biol. 2010, 397, 202–218.
- Furukawa, K.; Matsuzaki-Kobayashi, M.; Hasegawa, T.; Kikuchi, A.; Sugeno, N.; Itoyama, Y.; Wang, Y.; Yao, P.J.; Bushlin, I.; Takeda, A. Plasma membrane ion permeability induced by mutant alpha-synuclein contributes to the degeneration of neural cells. J. Neurochem. 2006, 97, 1071–1077.
- Feng, L.R.; Federoff, H.J.; Vicini, S.; Maguire-Zeiss, K.A. Alpha-synuclein mediates alterations in membrane conductance: A potential role for alpha-synuclein oligomers in cell vulnerability. Eur. J. Neurosci. 2010, 32, 10–17.
- Schmidt, F.; Levin, J.; Kamp, F.; Kretzschmar, H.; Giese, A.; Botzel, K. Single-channel electrophysiology reveals a distinct and uniform pore complex formed by alpha-synuclein oligomers in lipid membranes. PLoS ONE 2012, 7, e42545.
- Mironov, S.L. alpha-Synuclein forms non-selective cation channels and stimulates ATP-sensitive potassium channels in hippocampal neurons. J. Physiol. 2015, 593, 145–159.
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Lena, C.; et al. Alpha-synuclein assemblies sequester neuronal alpha3-Na+/K+-ATPase and impair Na+ gradient. EMBO J. 2015, 34, 2408–2423.
- Ronzitti, G.; Bucci, G.; Emanuele, M.; Leo, D.; Sotnikova, T.D.; Mus, L.V.; Soubrane, C.H.; Dallas, M.L.; Thalhammer, A.; Cingolani, L.A.; et al. Exogenous alpha-synuclein decreases raft partitioning of Cav2.2 channels inducing dopamine release. J. Neurosci. 2014, 34, 10603–10615.
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606.
- Pasternak, B.; Svanstrom, H.; Nielsen, N.M.; Fugger, L.; Melbye, M.; Hviid, A. Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol. 2012, 175, 627–635.
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086.
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232.
- McGranahan, T.M.; Patzlaff, N.E.; Grady, S.R.; Heinemann, S.F.; Booker, T.K. alpha4beta2 nicotinic acetylcholine receptors on dopaminergic neurons mediate nicotine reward and anxiety relief. J. Neurosci. 2011, 31, 10891–10902.
- Swant, J.; Goodwin, J.S.; North, A.; Ali, A.A.; Gamble-George, J.; Chirwa, S.; Khoshbouei, H. alpha-Synuclein stimulates a dopamine transporter-dependent chloride current and modulates the activity of the transporter. J. Biol. Chem. 2011, 286, 43933–43943.
- Butler, B.; Saha, K.; Rana, T.; Becker, J.P.; Sambo, D.; Davari, P.; Goodwin, J.S.; Khoshbouei, H. Dopamine transporter activity is modulated by alpha-synuclein. J. Biol. Chem. 2015, 290, 29542–29554.
- Sidhu, A.; Wersinger, C.; Vernier, P. alpha-Synuclein regulation of the dopaminergic transporter: A possible role in the pathogenesis of Parkinson’s disease. FEBS Lett. 2004, 565, 1–5.
- Dean, E.D.; Li, Y.; Torres, G.E.; Miller, G.W. Identification of a novel interaction between α-synuclein and VMAT2. FASEB J. 2008, 22, 715.6.
- Guo, J.T.; Chen, A.Q.; Kong, Q.; Zhu, H.; Ma, C.M.; Qin, C. Inhibition of vesicular monoamine transporter-2 activity in alpha-synuclein stably transfected SH-SY5Y cells. Cell Mol. Neurobiol. 2008, 28, 35–47.
- Perez, R.G.; Waymire, J.C.; Lin, E.; Liu, J.J.; Guo, F.; Zigmond, M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002, 22, 3090–3099.
- Baptista, M.J.; O’Farrell, C.; Daya, S.; Ahmad, R.; Miller, D.W.; Hardy, J.; Farrer, M.J.; Cookson, M.R. Co-ordinate transcriptional regulation of dopamine synthesis genes by alpha-synuclein in human neuroblastoma cell lines. J. Neurochem. 2003, 85, 957–968.
- Yu, S.; Zuo, X.; Li, Y.; Zhang, C.; Zhou, M.; Zhang, Y.A.; Ueda, K.; Chan, P. Inhibition of tyrosine hydroxylase expression in alpha-synuclein-transfected dopaminergic neuronal cells. Neurosci. Lett. 2004, 367, 34–39.
- Li, Y.H.; Gao, N.; Ye, Y.W.; Li, X.; Yu, S.; Yang, H.; Ueda, K.; Chan, P. Alpha-synuclein functions as a negative regulator for expression of tyrosine hydroxylase. Acta Neurol. Belg. 2011, 111, 130–135.
- Liu, D.; Jin, L.; Wang, H.; Zhao, H.; Zhao, C.; Duan, C.; Lu, L.; Wu, B.; Yu, S.; Chan, P.; et al. Silencing alpha-synuclein gene expression enhances tyrosine hydroxylase activity in MN9D cells. Neurochem. Res. 2008, 33, 1401–1409.
- Peng, X.; Tehranian, R.; Dietrich, P.; Stefanis, L.; Perez, R.G. Alpha-synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J. Cell Sci. 2005, 118, 3523–3530.
- Alerte, T.N.; Akinfolarin, A.A.; Friedrich, E.E.; Mader, S.A.; Hong, C.S.; Perez, R.G. Alpha-synuclein aggregation alters tyrosine hydroxylase phosphorylation and immunoreactivity: Lessons from viral transduction of knockout mice. Neurosci. Lett. 2008, 435, 24–29.
- Lou, H.; Montoya, S.E.; Alerte, T.N.; Wang, J.; Wu, J.; Peng, X.; Hong, C.S.; Friedrich, E.E.; Mader, S.A.; Pedersen, C.J.; et al. Serine 129 phosphorylation reduces the ability of alpha-synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo. J. Biol. Chem. 2010, 285, 17648–17661.
- Chu, Y.; Kordower, J.H. Age-associated increases of alpha-synuclein in monkeys and humans are associated with nigrostriatal dopamine depletion: Is this the target for Parkinson’s disease? Neurobiol. Dis. 2007, 25, 134–149.
- Chadchankar, H.; Ihalainen, J.; Tanila, H.; Yavich, L. Decreased reuptake of dopamine in the dorsal striatum in the absence of alpha-synuclein. Brain Res. 2011, 1382, 37–44.
- Al-Wandi, A.; Ninkina, N.; Millership, S.; Williamson, S.J.; Jones, P.A.; Buchman, V.L. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol. Aging 2010, 31, 796–804.
- Robertson, D.C.; Schmidt, O.; Ninkina, N.; Jones, P.A.; Sharkey, J.; Buchman, V.L. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J. Neurochem. 2004, 89, 1126–1136.
- Shen, J.; Du, T.; Wang, X.; Duan, C.; Gao, G.; Zhang, J.; Lu, L.; Yang, H. alpha-Synuclein amino terminus regulates mitochondrial membrane permeability. Brain Res. 2014, 1591, 14–26.
- Nakamura, K.; Nemani, V.M.; Azarbal, F.; Skibinski, G.; Levy, J.M.; Egami, K.; Munishkina, L.; Zhang, J.; Gardner, B.; Wakabayashi, J.; et al. Direct membrane association drives mitochondrial fission by the Parkinson disease-associated protein alpha-synuclein. J. Biol. Chem. 2011, 286, 20710–20726.
- Robotta, M.; Gerding, H.R.; Vogel, A.; Hauser, K.; Schildknecht, S.; Karreman, C.; Leist, M.; Subramaniam, V.; Drescher, M. Alpha-synuclein binds to the inner membrane of mitochondria in an alpha-helical conformation. ChemBioChem 2014, 15, 2499–2502.
- Faustini, G.; Marchesan, E.; Zonta, L.; Bono, F.; Bottani, E.; Longhena, F.; Ziviani, E.; Valerio, A.; Bellucci, A. Alpha-synuclein preserves mitochondrial fusion and function in neuronal cells. Oxid. Med. Cell Longev. 2019, 2019, 4246350.
- Ellis, C.E.; Murphy, E.J.; Mitchell, D.C.; Golovko, M.Y.; Scaglia, F.; Barcelo-Coblijn, G.C.; Nussbaum, R.L. Mitochondrial lipid abnormality and electron transport chain impairment in mice lacking alpha-synuclein. Mol. Cell Biol. 2005, 25, 10190–10201.
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric alpha-synuclein exerts a physiological role on brain ATP synthase. J. Neurosci. 2016, 36, 10510–10521.
- Hsu, L.J.; Sagara, Y.; Arroyo, A.; Rockenstein, E.; Sisk, A.; Mallory, M.; Wong, J.; Takenouchi, T.; Hashimoto, M.; Masliah, E. Alpha-synuclein promotes mitochondrial deficit and oxidative stress. Am. J. Pathol. 2000, 157, 401–410.
- Parihar, M.S.; Parihar, A.; Fujita, M.; Hashimoto, M.; Ghafourifar, P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol. Life Sci. 2008, 65, 1272–1284.
- Hu, D.; Sun, X.; Liao, X.; Zhang, X.; Zarabi, S.; Schimmer, A.; Hong, Y.; Ford, C.; Luo, Y.; Qi, X. Alpha-synuclein suppresses mitochondrial protease ClpP to trigger mitochondrial oxidative damage and neurotoxicity. Acta Neuropathol. 2019, 137, 939–960.
- Di Maio, R.; Barrett, P.J.; Hoffman, E.K.; Barrett, C.W.; Zharikov, A.; Borah, A.; Hu, X.; McCoy, J.; Chu, C.T.; Burton, E.A.; et al. Alpha-synuclein binds to TOM20 and inhibits mitochondrial protein import in Parkinson’s disease. Sci. Transl. Med. 2016, 8, 342ra78.
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100.
- Bir, A.; Sen, O.; Anand, S.; Khemka, V.K.; Banerjee, P.; Cappai, R.; Sahoo, A.; Chakrabarti, S. alpha-Synuclein-induced mitochondrial dysfunction in isolated preparation and intact cells: Implications in the pathogenesis of Parkinson’s disease. J. Neurochem. 2014, 131, 868–877.
- Perfeito, R.; Lazaro, D.F.; Outeiro, T.F.; Rego, A.C. Linking alpha-synuclein phosphorylation to reactive oxygen species formation and mitochondrial dysfunction in SH-SY5Y cells. Mol. Cell Neurosci. 2014, 62, 51–59.
- Subramaniam, S.R.; Vergnes, L.; Franich, N.R.; Reue, K.; Chesselet, M.F. Region specific mitochondrial impairment in mice with widespread overexpression of alpha-synuclein. Neurobiol. Dis. 2014, 70, 204–213.
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease alpha-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50.
- Fornai, F.; Schluter, O.M.; Lenzi, P.; Gesi, M.; Ruffoli, R.; Ferrucci, M.; Lazzeri, G.; Busceti, C.L.; Pontarelli, F.; Battaglia, G.; et al. Parkinson-like syndrome induced by continuous MPTP infusion: Convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc. Natl. Acad. Sci. USA 2005, 102, 3413–3418.
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644.
- Radad, K.; Al-Shraim, M.; Al-Emam, A.; Wang, F.; Kranner, B.; Rausch, W.D.; Moldzio, R. Rotenone: From modelling to implication in Parkinson’s disease. Folia Neuropathol. 2019, 57, 317–326.
- Vila, M.; Vukosavic, S.; Jackson-Lewis, V.; Neystat, M.; Jakowec, M.; Przedborski, S. Alpha-synuclein up-regulation in substantia nigra dopaminergic neurons following administration of the parkinsonian toxin MPTP. J. Neurochem. 2000, 74, 721–729.
- Chinta, S.J.; Mallajosyula, J.K.; Rane, A.; Andersen, J.K. Mitochondrial alpha-synuclein accumulation impairs complex I function in dopaminergic neurons and results in increased mitophagy in vivo. Neurosci. Lett. 2010, 486, 235–239.
- Loeb, V.; Yakunin, E.; Saada, A.; Sharon, R. The transgenic overexpression of alpha-synuclein and not its related pathology associates with complex I inhibition. J. Biol. Chem. 2010, 285, 7334–7343.
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507.
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca(2+) homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149.
- Chen, L.; Xie, Z.; Turkson, S.; Zhuang, X. A53T human alpha-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration. J. Neurosci. 2015, 35, 890–905.
- Poon, H.F.; Frasier, M.; Shreve, N.; Calabrese, V.; Wolozin, B.; Butterfield, D.A. Mitochondrial associated metabolic proteins are selectively oxidized in A30P alpha-synuclein transgenic mice—A model of familial Parkinson’s disease. Neurobiol. Dis. 2005, 18, 492–498.
- Kamp, F.; Exner, N.; Lutz, A.K.; Wender, N.; Hegermann, J.; Brunner, B.; Nuscher, B.; Bartels, T.; Giese, A.; Beyer, K.; et al. Inhibition of mitochondrial fusion by alpha-synuclein is rescued by PINK1, Parkin and DJ-1. EMBO J. 2010, 29, 3571–3589.
- Betarbet, R.; Sherer, T.B.; MacKenzie, G.; Garcia-Osuna, M.; Panov, A.V.; Greenamyre, J.T. Chronic systemic pesticide exposure reproduces features of Parkinson’s disease. Nat. Neurosci. 2000, 3, 1301–1306.
- Nistico, R.; Mehdawy, B.; Piccirilli, S.; Mercuri, N. Paraquat- and rotenone-induced models of Parkinson’s disease. Int. J. Immunopathol. Pharmacol. 2011, 24, 313–322.
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316.
- Lee, H.J.; Shin, S.Y.; Choi, C.; Lee, Y.H.; Lee, S.J. Formation and removal of alpha-synuclein aggregates in cells exposed to mitochondrial inhibitors. J. Biol. Chem. 2002, 277, 5411–5417.
- Kowall, N.W.; Hantraye, P.; Brouillet, E.; Beal, M.F.; McKee, A.C.; Ferrante, R.J. MPTP induces alpha-synuclein aggregation in the substantia nigra of baboons. Neuroreport 2000, 11, 211–213.
- Przedborski, S.; Chen, Q.; Vila, M.; Giasson, B.I.; Djaldatti, R.; Vukosavic, S.; Souza, J.M.; Jackson-Lewis, V.; Lee, V.M.; Ischiropoulos, H. Oxidative post-translational modifications of alpha-synuclein in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) mouse model of Parkinson’s disease. J. Neurochem. 2001, 76, 637–640.
- Hu, S.; Hu, M.; Liu, J.; Zhang, B.; Zhang, Z.; Zhou, F.H.; Wang, L.; Dong, J. Phosphorylation of tau and alpha-synuclein induced neurodegeneration in MPTP mouse model of Parkinson’s disease. Neuropsychiatry Dis. Treat. 2020, 16, 651–663.
- Lee, S.; Oh, S.T.; Jeong, H.J.; Pak, S.C.; Park, H.J.; Kim, J.; Cho, H.S.; Jeon, S. MPTP-induced vulnerability of dopamine neurons in A53T alpha-synuclein overexpressed mice with the potential involvement of DJ-1 downregulation. Korean J. Physiol. Pharmacol. 2017, 21, 625–632.
- Bazzu, G.; Calia, G.; Puggioni, G.; Spissu, Y.; Rocchitta, G.; Debetto, P.; Grigoletto, J.; Zusso, M.; Migheli, R.; Serra, P.A.; et al. alpha-Synuclein- and MPTP-generated rodent models of Parkinson’s disease and the study of extracellular striatal dopamine dynamics: A microdialysis approach. CNS Neurol. Disord. Drug Targets 2010, 9, 482–490.
- Grunewald, A.; Arns, B.; Seibler, P.; Rakovic, A.; Munchau, A.; Ramirez, A.; Sue, C.M.; Klein, C. ATP13A2 mutations impair mitochondrial function in fibroblasts from patients with Kufor-Rakeb syndrome. Neurobiol. Aging 2012, 33, 1843.e1.
- Park, J.S.; Koentjoro, B.; Veivers, D.; Mackay-Sim, A.; Sue, C.M. Parkinson’s disease-associated human ATP13A2 (PARK9) deficiency causes zinc dyshomeostasis and mitochondrial dysfunction. Hum. Mol. Genet. 2014, 23, 2802–2815.
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929.
- Guardia-Laguarta, C.; Area-Gomez, E.; Schon, E.A.; Przedborski, S. A new role for alpha-synuclein in Parkinson’s disease: Alteration of ER-mitochondrial communication. Mov. Disord. 2015, 30, 1026–1033.
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259.
- Jacobs, D.; Hoogerheide, D.P.; Rovini, A.; Jiang, Z.; Lee, J.C.; Rostovtseva, T.K.; Bezrukov, S.M. Probing membrane association of alpha-synuclein domains with VDAC nanopore reveals unexpected binding pattern. Sci. Rep. 2019, 9, 4580.
- Rostovtseva, T.K.; Gurnev, P.A.; Protchenko, O.; Hoogerheide, D.P.; Yap, T.L.; Philpott, C.C.; Lee, J.C.; Bezrukov, S.M. Alpha-synuclein shows high affinity interaction with voltage-dependent anion channel, suggesting mechanisms of mitochondrial regulation and toxicity in Parkinson disease. J. Biol. Chem. 2015, 290, 18467–18477.
- Hoogerheide, D.P.; Gurnev, P.A.; Rostovtseva, T.K.; Bezrukov, S.M. Mechanism of alpha-synuclein translocation through a VDAC nanopore revealed by energy landscape modeling of escape time distributions. Nanoscale 2017, 9, 183–192.
- Lu, L.; Zhang, C.; Cai, Q.; Lu, Q.; Duan, C.; Zhu, Y.; Yang, H. Voltage-dependent anion channel involved in the alpha-synuclein-induced dopaminergic neuron toxicity in rats. Acta Biochim. Biophys. Sin. 2013, 45, 170–178.
- Shoshan-Barmatz, V.; Zalk, R.; Gincel, D.; Vardi, N. Subcellular localization of VDAC in mitochondria and ER in the cerebellum. Biochim. Biophys. Acta 2004, 1657, 105–114.
- Shoshan-Barmatz, V.; Israelson, A. The voltage-dependent anion channel in endoplasmic/sarcoplasmic reticulum: Characterization, modulation and possible function. J. Membr. Biol. 2005, 204, 57–66.
- De Stefani, D.; Bononi, A.; Romagnoli, A.; Messina, A.; De Pinto, V.; Pinton, P.; Rizzuto, R. VDAC1 selectively transfers apoptotic Ca2+ signals to mitochondria. Cell Death Differ. 2012, 19, 267–273.
- Chu, Y.; Goldman, J.G.; Kelly, L.; He, Y.; Waliczek, T.; Kordower, J.H. Abnormal alpha-synuclein reduces nigral voltage-dependent anion channel 1 in sporadic and experimental Parkinson’s disease. Neurobiol. Dis. 2014, 69, 1–14.
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. Alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293.
- Martin, L.J.; Semenkow, S.; Hanaford, A.; Wong, M. Mitochondrial permeability transition pore regulates Parkinson’s disease development in mutant alpha-synuclein transgenic mice. Neurobiol. Aging 2014, 35, 1132–1152.
- Heman-Ackah, S.M.; Manzano, R.; Hoozemans, J.J.M.; Scheper, W.; Flynn, R.; Haerty, W.; Cowley, S.A.; Bassett, A.R.; Wood, M.J.A. Alpha-synuclein induces the unfolded protein response in Parkinson’s disease SNCA triplication iPSC-derived neurons. Hum. Mol. Genet. 2017, 26, 4441–4450.
- Lee, A.S. The ER chaperone and signaling regulator GRP78/BiP as a monitor of endoplasmic reticulum stress. Methods 2005, 35, 373–381.
- Wang, M.; Wey, S.; Zhang, Y.; Ye, R.; Lee, A.S. Role of the unfolded protein response regulator GRP78/BiP in development, cancer, and neurological disorders. Antioxid. Redox. Signal. 2009, 11, 2307–2316.
- Bellucci, A.; Navarria, L.; Zaltieri, M.; Falarti, E.; Bodei, S.; Sigala, S.; Battistin, L.; Spillantini, M.; Missale, C.; Spano, P. Induction of the unfolded protein response by alpha-synuclein in experimental models of Parkinson’s disease. J. Neurochem. 2011, 116, 588–605.
- Colla, E.; Coune, P.; Liu, Y.; Pletnikova, O.; Troncoso, J.C.; Iwatsubo, T.; Schneider, B.L.; Lee, M.K. Endoplasmic reticulum stress is important for the manifestations of alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3306–3320.
- Colla, E.; Jensen, P.H.; Pletnikova, O.; Troncoso, J.C.; Glabe, C.; Lee, M.K. Accumulation of toxic alpha-synuclein oligomer within endoplasmic reticulum occurs in alpha-synucleinopathy in vivo. J. Neurosci. 2012, 32, 3301–3305.
- Baek, J.H.; Whitfield, D.; Howlett, D.; Francis, P.; Bereczki, E.; Ballard, C.; Hortobagyi, T.; Attems, J.; Aarsland, D. Unfolded protein response is activated in Lewy body dementias. Neuropathol. Appl. Neurobiol. 2016, 42, 352–365.
- Yagi-Utsumi, M.; Satoh, T.; Kato, K. Structural basis of redox-dependent substrate binding of protein disulfide isomerase. Sci. Rep. 2015, 5, 13909.
- Honjo, Y.; Ito, H.; Horibe, T.; Takahashi, R.; Kawakami, K. Protein disulfide isomerase immunopositive glial cytoplasmic inclusions in patients with multiple system atrophy. Int. J. Neurosci. 2011, 121, 543–550.
- Conn, K.J.; Gao, W.; McKee, A.; Lan, M.S.; Ullman, M.D.; Eisenhauer, P.B.; Fine, R.E.; Wells, J.M. Identification of the protein disulfide isomerase family member PDIp in experimental Parkinson’s disease and Lewy body pathology. Brain Res. 2004, 1022, 164–172.
- Jin, J.; Li, G.J.; Davis, J.; Zhu, D.; Wang, Y.; Pan, C.; Zhang, J. Identification of novel proteins associated with both alpha-synuclein and DJ-1. Mol. Cell Proteom. 2007, 6, 845–859.
- Koch, G.; Smith, M.; Macer, D.; Webster, P.; Mortara, R. Endoplasmic reticulum contains a common, abundant calcium-binding glycoprotein, endoplasmin. J. Cell Sci. 1986, 86, 217–232.
- Labrador-Garrido, A.; Cejudo-Guillen, M.; Daturpalli, S.; Leal, M.M.; Klippstein, R.; De Genst, E.J.; Villadiego, J.; Toledo-Aral, J.J.; Dobson, C.M.; Jackson, S.E.; et al. Chaperome screening leads to identification of Grp94/Gp96 and FKBP4/52 as modulators of the alpha-synuclein-elicited immune response. FASEB J. 2016, 30, 564–577.
- Lee, A.S. Mammalian stress response: Induction of the glucose-regulated protein family. Curr. Opin. Cell Biol. 1992, 4, 267–273.
- Cooper, A.A.; Gitler, A.D.; Cashikar, A.; Haynes, C.M.; Hill, K.J.; Bhullar, B.; Liu, K.; Xu, K.; Strathearn, K.E.; Liu, F.; et al. Alpha-synuclein blocks ER-Golgi traffic and Rab1 rescues neuron loss in Parkinson’s models. Science 2006, 313, 324–328.
- Thayanidhi, N.; Helm, J.R.; Nycz, D.C.; Bentley, M.; Liang, Y.; Hay, J.C. Alpha-synuclein delays endoplasmic reticulum (ER)-to-Golgi transport in mammalian cells by antagonizing ER/Golgi SNAREs. Mol. Biol. Cell 2010, 21, 1850–1863.
- Gitler, A.D.; Bevis, B.J.; Shorter, J.; Strathearn, K.E.; Hamamichi, S.; Su, L.J.; Caldwell, K.A.; Caldwell, G.A.; Rochet, J.C.; McCaffery, J.M.; et al. The Parkinson’s disease protein alpha-synuclein disrupts cellular Rab homeostasis. Proc. Natl. Acad. Sci. USA 2008, 105, 145–150.
- Dalfo, E.; Gomez-Isla, T.; Rosa, J.L.; Nieto Bodelon, M.; Cuadrado Tejedor, M.; Barrachina, M.; Ambrosio, S.; Ferrer, I. Abnormal alpha-synuclein interactions with Rab proteins in alpha-synuclein A30P transgenic mice. J. Neuropathol. Exp. Neurol. 2004, 63, 302–313.
- Yin, G.; Lopes da Fonseca, T.; Eisbach, S.E.; Anduaga, A.M.; Breda, C.; Orcellet, M.L.; Szego, E.M.; Guerreiro, P.; Lazaro, D.F.; Braus, G.H.; et al. alpha-Synuclein interacts with the switch region of Rab8a in a Ser129 phosphorylation-dependent manner. Neurobiol. Dis. 2014, 70, 149–161.
- Breda, C.; Nugent, M.L.; Estranero, J.G.; Kyriacou, C.P.; Outeiro, T.F.; Steinert, J.R.; Giorgini, F. Rab11 modulates alpha-synuclein-mediated defects in synaptic transmission and behaviour. Hum. Mol. Genet. 2015, 24, 1077–1091.
- Fujita, Y.; Ohama, E.; Takatama, M.; Al-Sarraj, S.; Okamoto, K. Fragmentation of Golgi apparatus of nigral neurons with alpha-synuclein-positive inclusions in patients with Parkinson’s disease. Acta Neuropathol. 2006, 112, 261–265.
- Lee, H.J.; Khoshaghideh, F.; Lee, S.; Lee, S.J. Impairment of microtubule-dependent trafficking by overexpression of alpha-synuclein. Eur. J. Neurosci. 2006, 24, 3153–3162.
- Gosavi, N.; Lee, H.J.; Lee, J.S.; Patel, S.; Lee, S.J. Golgi fragmentation occurs in the cells with prefibrillar alpha-synuclein aggregates and precedes the formation of fibrillar inclusion. J. Biol. Chem. 2002, 277, 48984–48992.
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Cali, T.; Gai, W.P.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19.
- Wuytack, F.; Raeymaekers, L.; Missiaen, L. PMR1/SPCA Ca2+ pumps and the role of the Golgi apparatus as a Ca2+ store. Pflugers Arch. 2003, 446, 148–153.
- Buttner, S.; Faes, L.; Reichelt, W.N.; Broeskamp, F.; Habernig, L.; Benke, S.; Kourtis, N.; Ruli, D.; Carmona-Gutierrez, D.; Eisenberg, T.; et al. The Ca2+/Mn2+ ion-pump PMR1 links elevation of cytosolic Ca(2+) levels to alpha-synuclein toxicity in Parkinson’s disease models. Cell Death Differ. 2013, 20, 465–477.
- Nikoletopoulou, V.; Tavernarakis, N. The PMR1 pump in alpha-synuclein toxicity and neurodegeneration. Neurosci. Lett. 2018, 663, 66–71.
- Goers, J.; Manning-Bog, A.B.; McCormack, A.L.; Millett, I.S.; Doniach, S.; Di Monte, D.A.; Uversky, V.N.; Fink, A.L. Nuclear localization of alpha-synuclein and its interaction with histones. Biochemistry 2003, 42, 8465–8471.
- Mori, F.; Tanji, K.; Yoshimoto, M.; Takahashi, H.; Wakabayashi, K. Immunohistochemical comparison of alpha- and beta-synuclein in adult rat central nervous system. Brain Res. 2002, 941, 118–126.
- Yu, S.; Li, X.; Liu, G.; Han, J.; Zhang, C.; Li, Y.; Xu, S.; Liu, C.; Gao, Y.; Yang, H.; et al. Extensive nuclear localization of alpha-synuclein in normal rat brain neurons revealed by a novel monoclonal antibody. Neuroscience 2007, 145, 539–555.
- Huang, Z.; Xu, Z.; Wu, Y.; Zhou, Y. Determining nuclear localization of alpha-synuclein in synuclein mouse brains. Neuroscience 2011, 199, 318–332.
- Kontopoulos, E.; Parvin, J.D.; Feany, M.B. Alpha-synuclein acts in the nucleus to inhibit histone acetylation and promote neurotoxicity. Hum. Mol. Genet. 2006, 15, 3012–3023.
- Ma, K.L.; Song, L.K.; Yuan, Y.H.; Zhang, Y.; Han, N.; Gao, K.; Chen, N.H. The nuclear accumulation of alpha-synuclein is mediated by importin alpha and promotes neurotoxicity by accelerating the cell cycle. Neuropharmacology 2014, 82, 132–142.
- Desplats, P.; Spencer, B.; Coffee, E.; Patel, P.; Michael, S.; Patrick, C.; Adame, A.; Rockenstein, E.; Masliah, E. Alpha-synuclein sequesters Dnmt1 from the nucleus: A novel mechanism for epigenetic alterations in Lewy body diseases. J. Biol. Chem. 2011, 286, 9031–9037.
- Outeiro, T.F.; Kontopoulos, E.; Altmann, S.M.; Kufareva, I.; Strathearn, K.E.; Amore, A.M.; Volk, C.B.; Maxwell, M.M.; Rochet, J.C.; McLean, P.J.; et al. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson’s disease. Science 2007, 317, 516–519.
- Davidi, D.; Schechter, M.; Elhadi, S.A.; Matatov, A.; Nathanson, L.; Sharon, R. Alpha-synuclein translocates to the nucleus to activate retinoic-acid-dependent gene transcription. iScience 2020, 23, 100910.
- Schell, H.; Hasegawa, T.; Neumann, M.; Kahle, P.J. Nuclear and neuritic distribution of serine-129 phosphorylated alpha-synuclein in transgenic mice. Neuroscience 2009, 160, 796–804.
- Pinho, R.; Paiva, I.; Jercic, K.G.; Fonseca-Ornelas, L.; Gerhardt, E.; Fahlbusch, C.; Garcia-Esparcia, P.; Kerimoglu, C.; Pavlou, M.A.S.; Villar-Pique, A.; et al. Nuclear localization and phosphorylation modulate pathological effects of alpha-synuclein. Hum. Mol. Genet. 2019, 28, 31–50.
- Ryu, S.B.I.; Liew, H. Sumoylated α-synuclein translocates into the nucleus by karyopherin α6. Mol. Cell. Toxicol. 2019, 15, 103–109.
- Chen, V.; Moncalvo, M.; Tringali, D.; Tagliafierro, L.; Shriskanda, A.; Ilich, E.; Dong, W.; Kantor, B.; Chiba-Falek, O. The mechanistic role of alpha-synuclein in the nucleus: Impaired nuclear function caused by familial Parkinson’s disease SNCA mutations. Hum. Mol. Genet. 2020, 29, 3107–3121.
- Schaser, A.J.; Osterberg, V.R.; Dent, S.E.; Stackhouse, T.L.; Wakeham, C.M.; Boutros, S.W.; Weston, L.J.; Owen, N.; Weissman, T.A.; Luna, E.; et al. Alpha-synuclein is a DNA binding protein that modulates DNA repair with implications for Lewy body disorders. Sci. Rep. 2019, 9, 10919.
- Liu, X.; Lee, Y.J.; Liou, L.C.; Ren, Q.; Zhang, Z.; Wang, S.; Witt, S.N. Alpha-synuclein functions in the nucleus to protect against hydroxyurea-induced replication stress in yeast. Hum. Mol. Genet. 2011, 20, 3401–3414.
- Bennett, M.C.; Bishop, J.F.; Leng, Y.; Chock, P.B.; Chase, T.N.; Mouradian, M.M. Degradation of alpha-synuclein by proteasome. J. Biol. Chem. 1999, 274, 33855–33858.
- Webb, J.L.; Ravikumar, B.; Atkins, J.; Skepper, J.N.; Rubinsztein, D.C. Alpha-synuclein is degraded by both autophagy and the proteasome. J. Biol. Chem. 2003, 278, 25009–25013.
- Vogiatzi, T.; Xilouri, M.; Vekrellis, K.; Stefanis, L. Wild type alpha-synuclein is degraded by chaperone-mediated autophagy and macroautophagy in neuronal cells. J. Biol. Chem. 2008, 283, 23542–23556.
- Rott, R.; Szargel, R.; Haskin, J.; Bandopadhyay, R.; Lees, A.J.; Shani, V.; Engelender, S. alpha-Synuclein fate is determined by USP9X-regulated monoubiquitination. Proc. Natl. Acad. Sci. USA 2011, 108, 18666–18671.
- Haj-Yahya, M.; Fauvet, B.; Herman-Bachinsky, Y.; Hejjaoui, M.; Bavikar, S.N.; Karthikeyan, S.V.; Ciechanover, A.; Lashuel, H.A.; Brik, A. Synthetic polyubiquitinated alpha-Synuclein reveals important insights into the roles of the ubiquitin chain in regulating its pathophysiology. Proc. Natl. Acad. Sci. USA 2013, 110, 17726–17731.
- Tofaris, G.K.; Layfield, R.; Spillantini, M.G. alpha-synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Lett. 2001, 509, 22–26.
- Liu, C.W.; Corboy, M.J.; DeMartino, G.N.; Thomas, P.J. Endoproteolytic activity of the proteasome. Science 2003, 299, 408–411.
- Rideout, H.J.; Larsen, K.E.; Sulzer, D.; Stefanis, L. Proteasomal inhibition leads to formation of ubiquitin/alpha-synuclein-immunoreactive inclusions in PC12 cells. J. Neurochem. 2001, 78, 899–908.
- Rideout, H.J.; Stefanis, L. Proteasomal inhibition-induced inclusion formation and death in cortical neurons require transcription and ubiquitination. Mol. Cell. Neurosci. 2002, 21, 223–238.
- Emmanouilidou, E.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein oligomers are targeted to, and impair, the 26S proteasome. Neurobiol. Aging 2010, 31, 953–968.
- Ebrahimi-Fakhari, D.; Cantuti-Castelvetri, I.; Fan, Z.; Rockenstein, E.; Masliah, E.; Hyman, B.T.; McLean, P.J.; Unni, V.K. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of alpha-synuclein. J. Neurosci. 2011, 31, 14508–14520.
- Xilouri, M.; Brekk, O.R.; Stefanis, L. alpha-Synuclein and protein degradation systems: A reciprocal relationship. Mol. Neurobiol. 2013, 47, 537–551.
- Cuervo, A.M.; Stefanis, L.; Fredenburg, R.; Lansbury, P.T.; Sulzer, D. Impaired degradation of mutant alpha-synuclein by chaperone-mediated autophagy. Science 2004, 305, 1292–1295.
- Lee, H.J.; Khoshaghideh, F.; Patel, S.; Lee, S.J. Clearance of alpha-synuclein oligomeric intermediates via the lysosomal degradation pathway. J. Neurosci. 2004, 24, 1888–1896.
- Mak, S.K.; McCormack, A.L.; Manning-Bog, A.B.; Cuervo, A.M.; Di Monte, D.A. Lysosomal degradation of alpha-synuclein in vivo. J. Biol. Chem. 2010, 285, 13621–13629.
- Xilouri, M.; Brekk, O.R.; Landeck, N.; Pitychoutis, P.M.; Papasilekas, T.; Papadopoulou-Daifoti, Z.; Kirik, D.; Stefanis, L. Boosting chaperone-mediated autophagy in vivo mitigates alpha-synuclein-induced neurodegeneration. Brain 2013, 136, 2130–2146.
- Issa, A.R.; Sun, J.; Petitgas, C.; Mesquita, A.; Dulac, A.; Robin, M.; Mollereau, B.; Jenny, A.; Cherif-Zahar, B.; Birman, S. The lysosomal membrane protein LAMP2A promotes autophagic flux and prevents SNCA-induced Parkinson disease-like symptoms in the Drosophila brain. Autophagy 2018, 14, 1898–1910.
- Xilouri, M.; Brekk, O.R.; Polissidis, A.; Chrysanthou-Piterou, M.; Kloukina, I.; Stefanis, L. Impairment of chaperone-mediated autophagy induces dopaminergic neurodegeneration in rats. Autophagy 2016, 12, 2230–2247.
- Alvarez-Erviti, L.; Rodriguez-Oroz, M.C.; Cooper, J.M.; Caballero, C.; Ferrer, I.; Obeso, J.A.; Schapira, A.H. Chaperone-mediated autophagy markers in Parkinson disease brains. Arch. Neurol. 2010, 67, 1464–1472.
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Spiro, A.S.; Furuta, A.; Cooper, A.; Garner, B.; Kabuta, T.; Halliday, G.M. Lysosomal-associated membrane protein 2 isoforms are differentially affected in early Parkinson’s disease. Mov. Disord. 2015, 30, 1639–1647.
- Tanaka, Y.; Engelender, S.; Igarashi, S.; Rao, R.K.; Wanner, T.; Tanzi, R.E.; Sawa, A.; Valina, L.D.; Dawson, T.M.; Ross, C.A. Inducible expression of mutant alpha-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Hum. Mol. Genet. 2001, 10, 919–926.
- Stefanis, L.; Larsen, K.E.; Rideout, H.J.; Sulzer, D.; Greene, L.A. Expression of A53T mutant but not wild-type alpha-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. J. Neurosci. 2001, 21, 9549–9560.
- Martin-Clemente, B.; Alvarez-Castelao, B.; Mayo, I.; Sierra, A.B.; Diaz, V.; Milan, M.; Farinas, I.; Gomez-Isla, T.; Ferrer, I.; Castano, J.G. alpha-Synuclein expression levels do not significantly affect proteasome function and expression in mice and stably transfected PC12 cell lines. J. Biol. Chem. 2004, 279, 52984–52990.
- Zondler, L.; Kostka, M.; Garidel, P.; Heinzelmann, U.; Hengerer, B.; Mayer, B.; Weishaupt, J.H.; Gillardon, F.; Danzer, K.M. Proteasome impairment by alpha-synuclein. PLoS ONE 2017, 12, e0184040.
- Bentea, E.; Verbruggen, L.; Massie, A. The proteasome inhibition model of Parkinson’s disease. J. Parkinsons Dis. 2017, 7, 31–63.
- McNaught, K.S.; Jenner, P. Proteasomal function is impaired in substantia nigra in Parkinson’s disease. Neurosci. Lett. 2001, 297, 191–194.
- McNaught, K.S.; Belizaire, R.; Isacson, O.; Jenner, P.; Olanow, C.W. Altered proteasomal function in sporadic Parkinson’s disease. Exp. Neurol. 2003, 179, 38–46.
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633.
- Winslow, A.R.; Chen, C.W.; Corrochano, S.; Acevedo-Arozena, A.; Gordon, D.E.; Peden, A.A.; Lichtenberg, M.; Menzies, F.M.; Ravikumar, B.; Imarisio, S.; et al. Alpha-synuclein impairs macroautophagy: Implications for Parkinson’s disease. J. Cell Biol. 2010, 190, 1023–1037.
- Zavodszky, E.; Seaman, M.N.; Moreau, K.; Jimenez-Sanchez, M.; Breusegem, S.Y.; Harbour, M.E.; Rubinsztein, D.C. Mutation in VPS35 associated with Parkinson’s disease impairs WASH complex association and inhibits autophagy. Nat. Commun. 2014, 5, 3828.
- Yan, J.Q.; Yuan, Y.H.; Gao, Y.N.; Huang, J.Y.; Ma, K.L.; Gao, Y.; Zhang, W.Q.; Guo, X.F.; Chen, N.H. Overexpression of human E46K mutant alpha-synuclein impairs macroautophagy via inactivation of JNK1-Bcl-2 pathway. Mol. Neurobiol. 2014, 50, 685–701.
- Lei, Z.; Cao, G.; Wei, G. A30P mutant alpha-synuclein impairs autophagic flux by inactivating JNK signaling to enhance ZKSCAN3 activity in midbrain dopaminergic neurons. Cell Death Dis 2019, 10, 133.
- Choubey, V.; Safiulina, D.; Vaarmann, A.; Cagalinec, M.; Wareski, P.; Kuum, M.; Zharkovsky, A.; Kaasik, A. Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy. J. Biol. Chem. 2011, 286, 10814–10824.
- Martinez-Vicente, M.; Talloczy, Z.; Kaushik, S.; Massey, A.C.; Mazzulli, J.; Mosharov, E.V.; Hodara, R.; Fredenburg, R.; Wu, D.C.; Follenzi, A.; et al. Dopamine-modified alpha-synuclein blocks chaperone-mediated autophagy. J. Clin. Investig. 2008, 118, 777–788.
- Mazzulli, J.R.; Zunke, F.; Isacson, O.; Studer, L.; Krainc, D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc. Natl. Acad. Sci. USA 2016, 113, 1931–1936.
- Osellame, L.D.; Rahim, A.A.; Hargreaves, I.P.; Gegg, M.E.; Richard-Londt, A.; Brandner, S.; Waddington, S.N.; Schapira, A.H.V.; Duchen, M.R. Mitochondria and quality control defects in a mouse model of Gaucher disease—Links to Parkinson’s disease. Cell Metab. 2013, 17, 941–953.
- Sardi, S.P.; Clarke, J.; Viel, C.; Chan, M.; Tamsett, T.J.; Treleaven, C.M.; Bu, J.; Sweet, L.; Passini, M.A.; Dodge, J.C.; et al. Augmenting CNS glucocerebrosidase activity as a therapeutic strategy for parkinsonism and other Gaucher-related synucleinopathies. Proc. Natl. Acad. Sci. USA 2013, 110, 3537–3542.
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848.
- Du, T.T.; Wang, L.; Duan, C.L.; Lu, L.L.; Zhang, J.L.; Gao, G.; Qiu, X.B.; Wang, X.M.; Yang, H. GBA deficiency promotes SNCA/alpha-synuclein accumulation through autophagic inhibition by inactivated PPP2A. Autophagy 2015, 11, 1803–1820.
- Liu, G.; Chen, M.; Mi, N.; Yang, W.; Li, X.; Wang, P.; Yin, N.; Li, Y.; Yue, F.; Chan, P.; et al. Increased oligomerization and phosphorylation of alpha-synuclein are associated with decreased activity of glucocerebrosidase and protein phosphatase 2A in aging monkey brains. Neurobiol. Aging 2015, 36, 2649–2659.
- Rocha, E.M.; Smith, G.A.; Park, E.; Cao, H.; Graham, A.R.; Brown, E.; McLean, J.R.; Hayes, M.A.; Beagan, J.; Izen, S.C.; et al. Sustained systemic glucocerebrosidase inhibition induces brain alpha-synuclein aggregation, microglia and complement C1q activation in mice. Antioxid. Redox. Signal. 2015, 23, 550–564.
- Borghi, R.; Marchese, R.; Negro, A.; Marinelli, L.; Forloni, G.; Zaccheo, D.; Abbruzzese, G.; Tabaton, M. Full length alpha-synuclein is present in cerebrospinal fluid from Parkinson’s disease and normal subjects. Neurosci. Lett. 2000, 287, 65–67.
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Cooper, L.J.; Fullwood, N.J.; Gibson, M.J.; Curran, M.D.; Court, J.A.; Mann, D.M.; Ikeda, S.; et al. Alpha-synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. FASEB J. 2003, 17, 1945–1947.
- Lee, H.J.; Patel, S.; Lee, S.J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024.
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851.
- Danzer, K.M.; Kranich, L.R.; Ruf, W.P.; Cagsal-Getkin, O.; Winslow, A.R.; Zhu, L.; Vanderburg, C.R.; McLean, P.J. Exosomal cell-to-cell transmission of alpha synuclein oligomers. Mol. Neurodegener. 2012, 7, 42.
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506.
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503.
- Li, J.Y.; Englund, E.; Widner, H.; Rehncrona, S.; Bjorklund, A.; Lindvall, O.; Brundin, P. Characterization of Lewy body pathology in 12- and 16-year-old intrastriatal mesencephalic grafts surviving in a patient with Parkinson’s disease. Mov. Disord. 2010, 25, 1091–1096.
- Kordower, J.H.; Dodiya, H.B.; Kordower, A.M.; Terpstra, B.; Paumier, K.; Madhavan, L.; Sortwell, C.; Steece-Collier, K.; Collier, T.J. Transfer of host-derived alpha synuclein to grafted dopaminergic neurons in rat. Neurobiol. Dis. 2011, 43, 552–557.
- Kurowska, Z.; Englund, E.; Widner, H.; Lindvall, O.; Li, J.Y.; Brundin, P. Signs of degeneration in 12–22-year old grafts of mesencephalic dopamine neurons in patients with Parkinson’s disease. J. Parkinsons Dis. 2011, 1, 83–92.
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Oikawa, T.; Arai, T.; Akiyama, H.; Mann, D.M.; Hasegawa, M. Prion-like spreading of pathological alpha-synuclein in brain. Brain 2013, 136, 1128–1138.
- Recasens, A.; Dehay, B.; Bove, J.; Carballo-Carbajal, I.; Dovero, S.; Perez-Villalba, A.; Fernagut, P.O.; Blesa, J.; Parent, A.; Perier, C.; et al. Lewy body extracts from Parkinson disease brains trigger alpha-synuclein pathology and neurodegeneration in mice and monkeys. Ann. Neurol. 2014, 75, 351–362.
- Luk, K.C.; Kehm, V.; Carroll, J.; Zhang, B.; O’Brien, P.; Trojanowski, J.Q.; Lee, V.M. Pathological alpha-synuclein transmission initiates Parkinson-like neurodegeneration in nontransgenic mice. Science 2012, 338, 949–953.
- Luk, K.C.; Song, C.; O’Brien, P.; Stieber, A.; Branch, J.R.; Brunden, K.R.; Trojanowski, J.Q.; Lee, V.M. Exogenous alpha-synuclein fibrils seed the formation of Lewy body-like intracellular inclusions in cultured cells. Proc. Natl. Acad. Sci. USA 2009, 106, 20051–20056.
- Sacino, A.N.; Brooks, M.; Thomas, M.A.; McKinney, A.B.; Lee, S.; Regenhardt, R.W.; McGarvey, N.H.; Ayers, J.I.; Notterpek, L.; Borchelt, D.R.; et al. Intramuscular injection of alpha-synuclein induces CNS alpha-synuclein pathology and a rapid-onset motor phenotype in transgenic mice. Proc. Natl. Acad. Sci. USA 2014, 111, 10732–10737.
- Paumier, K.L.; Luk, K.C.; Manfredsson, F.P.; Kanaan, N.M.; Lipton, J.W.; Collier, T.J.; Steece-Collier, K.; Kemp, C.J.; Celano, S.; Schulz, E.; et al. Intrastriatal injection of pre-formed mouse alpha-synuclein fibrils into rats triggers alpha-synuclein pathology and bilateral nigrostriatal degeneration. Neurobiol. Dis. 2015, 82, 185–199.
- Abdelmotilib, H.; Maltbie, T.; Delic, V.; Liu, Z.; Hu, X.; Fraser, K.B.; Moehle, M.S.; Stoyka, L.; Anabtawi, N.; Krendelchtchikova, V.; et al. alpha-Synuclein fibril-induced inclusion spread in rats and mice correlates with dopaminergic Neurodegeneration. Neurobiol. Dis. 2017, 105, 84–98.
- Gustafsson, G.; Loov, C.; Persson, E.; Lazaro, D.F.; Takeda, S.; Bergstrom, J.; Erlandsson, A.; Sehlin, D.; Balaj, L.; Gyorgy, B.; et al. Secretion and uptake of alpha-synuclein via extracellular vesicles in cultured cells. Cell. Mol. Neurobiol. 2018, 38, 1539–1550.
- Fussi, N.; Hollerhage, M.; Chakroun, T.; Nykanen, N.P.; Rosler, T.W.; Koeglsperger, T.; Wurst, W.; Behrends, C.; Hoglinger, G.U. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 2018, 9, 757.
- El-Agnaf, O.M.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of alpha-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425.
- Delenclos, M.; Trendafilova, T.; Mahesh, D.; Baine, A.M.; Moussaud, S.; Yan, I.K.; Patel, T.; McLean, P.J. Investigation of endocytic pathways for the internalization of exosome-associated oligomeric alpha-synuclein. Front. Neurosci. 2017, 11, 172.
- Sung, J.Y.; Kim, J.; Paik, S.R.; Park, J.H.; Ahn, Y.S.; Chung, K.C. Induction of neuronal cell death by Rab5A-dependent endocytosis of alpha-synuclein. J. Biol. Chem. 2001, 276, 27441–27448.
- Lee, H.J.; Suk, J.E.; Bae, E.J.; Lee, J.H.; Paik, S.R.; Lee, S.J. Assembly-dependent endocytosis and clearance of extracellular alpha-synuclein. Int. J. Biochem. Cell Biol. 2008, 40, 1835–1849.
- Ahn, K.J.; Paik, S.R.; Chung, K.C.; Kim, J. Amino acid sequence motifs and mechanistic features of the membrane translocation of alpha-synuclein. J. Neurochem. 2006, 97, 265–279.
- Abounit, S.; Bousset, L.; Loria, F.; Zhu, S.; de Chaumont, F.; Pieri, L.; Olivo-Marin, J.C.; Melki, R.; Zurzolo, C. Tunneling nanotubes spread fibrillar alpha-synuclein by intercellular trafficking of lysosomes. EMBO J. 2016, 35, 2120–2138.
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366.
- Jao, C.C.; Hegde, B.G.; Chen, J.; Haworth, I.S.; Langen, R. Structure of membrane-bound alpha-synuclein from site-directed spin labeling and computational refinement. Proc. Natl. Acad. Sci. USA 2008, 105, 19666–19671.
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.I.; Yin, X.; Xiong, Y.; Ge, P.; Umanah, G.E.; Brahmachari, S.; Shin, J.H.; et al. Pathological alpha-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353.
- Ihse, E.; Yamakado, H.; van Wijk, X.M.; Lawrence, R.; Esko, J.D.; Masliah, E. Cellular internalization of alpha-synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017, 7, 9008.
- Rey, N.L.; Steiner, J.A.; Maroof, N.; Luk, K.C.; Madaj, Z.; Trojanowski, J.Q.; Lee, V.M.; Brundin, P. Widespread transneuronal propagation of alpha-synucleinopathy triggered in olfactory bulb mimics prodromal Parkinson’s disease. J. Exp. Med. 2016, 213, 1759–1778.
- Karampetsou, M.; Ardah, M.T.; Semitekolou, M.; Polissidis, A.; Samiotaki, M.; Kalomoiri, M.; Majbour, N.; Xanthou, G.; El-Agnaf, O.M.A.; Vekrellis, K. Phosphorylated exogenous alpha-synuclein fibrils exacerbate pathology and induce neuronal dysfunction in mice. Sci. Rep. 2017, 7, 16533.
- Karpowicz, R.J., Jr.; Haney, C.M.; Mihaila, T.S.; Sandler, R.M.; Petersson, E.J.; Lee, V.M. Selective imaging of internalized proteopathic alpha-synuclein seeds in primary neurons reveals mechanistic insight into transmission of synucleinopathies. J. Biol. Chem. 2017, 292, 13482–13497.
- Luna, E.; Decker, S.C.; Riddle, D.M.; Caputo, A.; Zhang, B.; Cole, T.; Caswell, C.; Xie, S.X.; Lee, V.M.Y.; Luk, K.C. Differential alpha-synuclein expression contributes to selective vulnerability of hippocampal neuron subpopulations to fibril-induced toxicity. Acta Neuropathol. 2018, 135, 855–875.
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146.
- Sacino, A.N.; Brooks, M.; McKinney, A.B.; Thomas, M.A.; Shaw, G.; Golde, T.E.; Giasson, B.I. Brain injection of alpha-synuclein induces multiple proteinopathies, gliosis, and a neuronal injury marker. J. Neurosci. 2014, 34, 12368–12378.
- Wu, Q.; Takano, H.; Riddle, D.M.; Trojanowski, J.Q.; Coulter, D.A.; Lee, V.M. Alpha-synuclein (alphaSyn) preformed fibrils induce endogenous alphaSyn aggregation, compromise synaptic activity and enhance synapse loss in cultured excitatory hippocampal neurons. J. Neurosci. 2019, 39, 5080–5094.
- Chu, Y.; Kordower, J.H. The prion hypothesis of Parkinson’s disease. Curr. Neurol. Neurosci. Rep. 2015, 15, 28.
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744.
- Wood, S.J.; Wypych, J.; Steavenson, S.; Louis, J.C.; Citron, M.; Biere, A.L. alpha-synuclein fibrillogenesis is nucleation-dependent. Implications for the pathogenesis of Parkinson’s disease. J. Biol. Chem. 1999, 274, 19509–19512.
- Masuda-Suzukake, M.; Nonaka, T.; Hosokawa, M.; Kubo, M.; Shimozawa, A.; Akiyama, H.; Hasegawa, M. Pathological alpha-synuclein propagates through neural networks. Acta Neuropathol. Commun. 2014, 2, 88.
- Freundt, E.C.; Maynard, N.; Clancy, E.K.; Roy, S.; Bousset, L.; Sourigues, Y.; Covert, M.; Melki, R.; Kirkegaard, K.; Brahic, M. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann. Neurol. 2012, 72, 517–524.
- Mezias, C.; Rey, N.; Brundin, P.; Raj, A. Neural connectivity predicts spreading of alpha-synuclein pathology in fibril-injected mouse models: Involvement of retrograde and anterograde axonal propagation. Neurobiol. Dis. 2020, 134, 104623.
- Tang, Y.; Das, U.; Scott, D.A.; Roy, S. The slow axonal transport of alpha-synuclein--mechanistic commonalities amongst diverse cytosolic cargoes. Cytoskeleton 2012, 69, 506–513.
- Saha, A.R.; Hill, J.; Utton, M.A.; Asuni, A.A.; Ackerley, S.; Grierson, A.J.; Miller, C.C.; Davies, A.M.; Buchman, V.L.; Anderton, B.H.; et al. Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J. Cell Sci. 2004, 117, 1017–1024.
- Li, W.; Hoffman, P.N.; Stirling, W.; Price, D.L.; Lee, M.K. Axonal transport of human alpha-synuclein slows with aging but is not affected by familial Parkinson’s disease-linked mutations. J. Neurochem. 2004, 88, 401–410.
- Prots, I.; Veber, V.; Brey, S.; Campioni, S.; Buder, K.; Riek, R.; Bohm, K.J.; Winner, B. alpha-Synuclein oligomers impair neuronal microtubule-kinesin interplay. J. Biol. Chem. 2013, 288, 21742–21754.
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain 2012, 135, 2058–2073.
- Chung, C.Y.; Koprich, J.B.; Siddiqi, H.; Isacson, O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009, 29, 3365–3373.
- Holec, S.A.M.; Woerman, A.L. Evidence of distinct alpha-synuclein strains underlying disease heterogeneity. Acta Neuropathol. 2020.
- Lau, A.; So, R.W.L.; Lau, H.H.C.; Sang, J.C.; Ruiz-Riquelme, A.; Fleck, S.C.; Stuart, E.; Menon, S.; Visanji, N.P.; Meisl, G.; et al. alpha-Synuclein strains target distinct brain regions and cell types. Nat. Neurosci. 2020, 23, 21–31.
- Peelaerts, W.; Bousset, L.; Van der Perren, A.; Moskalyuk, A.; Pulizzi, R.; Giugliano, M.; Van den Haute, C.; Melki, R.; Baekelandt, V. Alpha-synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature 2015, 522, 340–344.
- Rey, N.L.; Bousset, L.; George, S.; Madaj, Z.; Meyerdirk, L.; Schulz, E.; Steiner, J.A.; Melki, R.; Brundin, P. alpha-Synuclein conformational strains spread, seed and target neuronal cells differentially after injection into the olfactory bulb. Acta Neuropathol. Commun. 2019, 7, 221.


