Immune activation in the central nervous system involves mostly microglia in response to pathogen invasion or tissue damage, which react, promoting a self-limiting inflammatory response aimed to restore homeostasis. However, prolonged, uncontrolled inflammation may result in the production by microglia of neurotoxic factors that lead to the amplification of the disease state and tissue damage. In particular, specific inducers of inflammation associated with neurodegenerative diseases activate inflammatory processes that result in the production of a number of mediators and cytokines that enhance neurodegenerative processes. Phosphoinositide 3-kinases (PI3Ks) constitute a family of enzymes regulating a wide range of activity, including signal transduction.
1. Introduction
Neurodegeneration, a slow and progressive destruction of neuronal cells, represents the pathological condition common to several brain disorders, including Alzheimer’s disease (AD), Parkinson’s disease (PD), multiple sclerosis (MS), Huntington’s disease (HD), and amyotrophic lateral sclerosis (ALS)
[1][2].
Among the many cellular and molecular processes responsible for neurodegenerative events, such as oxidative responses, accumulation of protein aggregates, altered mitochondrial function, and triggering of apoptosis
[2], neuroinflammation is correlated with the onset and progression of a number of neurodegenerative diseases of both an acute and chronic nature
[3][4][5]. In general terms, neuroinflammation is a defense mechanism aimed to protect the central nervous system (CNS) in response to a variety of insults, including infection, traumatic injury, toxic metabolites, or autoimmune events
[6]. Although it is believed that an acute neuroinflammatory response is generally gainful to the CNS, minimizing further injury and contributing to the tissue homeostasis, chronic neuroinflammation in the CNS can give rise to severe damage to the neuronal compartment, interfering with its homeostatic integrity, ruffling the balance between reparative responses and pro-inflammatory events
[7].
Multiple studies suggest the involvement of microglia as a critical executor of the inflammation mediated neurodegeneration. Microglia, the resident immune cells of the CNS, play a defense role in the CNS against potentially dangerous agents, although recently, an emerging role has been described concerning their involvement in the prompting of pathological processes. Active microglia have beneficial functions, promoting pathogen clearance and tissue regeneration, contributing to neuronal survival through the release of beneficial trophic factors
[8]. Interestingly, microglia are multifaceted cells since they may change their phenotype and function during neurodegenerative disorders, such as PD, AD, MS, ALS, stroke, and traumatic brain injury (TBI), eliciting pro-inflammatory responses
[8].
If left unchecked, during brain damage or infection, activated microglia prompt and sustain an inflammatory phenotype, releasing a variety of neurotoxic molecules, ultimately leading to neuronal demise
[9]. Although recent lines of evidence attributed to microglia a common disease associated signature
[10][11][12], the mechanisms regulating the microglial phenotype in neurodegenerative processes have not yet been clarified.
Phosphoinositide 3-kinases (PI3Ks) regulate several key mechanisms in the inflammatory response to external insults
[13]. PI3Ks are enzymes able to transduce signals deriving from growth factors, cytokines, hormones, as well as lipopolysaccharide (LPS) into intracellular messages, thus generating phospholipids. These, in turn, lead to the serine/threonine kinase Akt and other downstream effector pathways. Eight PI3Ks are described in mammalian cells and classified into three families
[14], differing in their regulation and preferred lipid substrate. The PI3K lipid kinases are involved in many cellular functions, including signal transduction, as well as intracellular vesicular traffic, then dysregulation of PI3K pathways is involved in a number of pathological conditions such as neurological diseases. Experimental evidence indicated that inhibition of the PI3K/Akt pathway in the LPS activated microglia leads to a reduced level of proinflammatory factors
[15]. Conversely, another study reported that PI3K/Akt activation contributes to the attenuation of brain damage, being able to upregulate anti-oxidative response with the inhibition of inflammation and apoptosis
[16]. Therefore, these results point out that there are diversified effects mediated by this crucial signal molecule in orchestrating both protective and possibly harmful responses.
2. PI3K Signaling Pathway
PI3Ks constitute a conserved family of kinases able to phosphorylate one or more inositol phospholipids in the 3-position present on the inositol ring, generating other molecules such as phosphatidylinositol-3,4,5-trisphosphate (PtdIns(3,4,5)P3,phosphatidylinositol-3,4-bisphosphate (PtdIns(3,4)P2), and phosphatidylinositol-3-phosphate (PtdIns(3)P)
[14][17]. These PI3K enzymes are located on the inner side of the plasma membrane where they propagate intracellular signaling cascades regulating a wide range of actions that comprise signal transduction, vesicular traffic, and cytoskeletal reorganization
[14][18].
2.1. Classes of PI3K Enzymes
In mammalian cells, the PI3Ks can be divided into three classes (I, II, and III) according to substrate affinity, functional homologies and their structural characteristics. Class I PI3Ks are composed of two subtypes, classes IA and IB, based on different associated adaptors
[19].
The heterodimers of class IA consist of a catalytic p110 and a p85 regulatory subunit. The p110 isoforms include three different types (110α, p110β, and p110δ), whereas there are five regulatory p85 polypeptides (p85α, p85β, p55α, p50α, and p55γ)
[20][21]. The p110δ isoform is mostly confined to expression in immune cells, whereas the p110α and β isoforms are expressed in all cell types
[22]. The p85α and β regulatory subunits are expressed in many cells, whereas the other polypeptides are distributed in a few tissues (see ).
Table 1. Tissue distribution of PI3K isoforms.
| PI3k |
Expression |
Ref |
| p110 α |
Ubiquitous |
[14][23] |
| p110 β |
Ubiquitous |
[14][23][24] |
| p110 δ |
Immune cells, Neurons and Microglia, Spleen, Platelets, Endothelial Cells |
[25] |
| p85 α |
Ubiquitous |
[24] |
| p55 α |
Brain, Muscle |
[26] |
| p50 α |
Liver, Kidney, Brain, T Cells |
[25] |
| p85 β |
Ubiquitous |
[24][26] |
| p55 γ |
Brain, Testis, Liver, Muscle, Fat, Spleen |
[24] |
| p110 γ |
Immune Cells, Heart, Pancreas, Liver, Skeletal Muscle |
[25][27] |
| p101 |
Immune Cells, Mast Cells |
[26][28][29] |
| p84/p87 |
Immune cells, Mast Cell, Heart |
[26][28][29] |
| pI3k-C2 |
Ubiquitous |
[30] |
| pI3k-C2β |
Ubiquitous |
[30] |
| pI3k-C2 γ |
Liver, Prostate, Breast, Salivary Gland |
[31] |
| Vps34 |
Ubiquitous |
[32] |
| Vps15 |
Ubiquitous |
[26] |
The p110 subunits contain five regions: a C-2 region for membrane anchoring, a Ras binding region (RBD), an N-terminal region called the adaptor binding region (ABD) that interacts with the regulatory p85 family members, a helical region, and a kinase catalytic region. The regulatory subunits share three core regions, including a p110 binding region called the inter-Src-homology-2 (iSH2) region, flanked by two SH2 regions that bind constitutively to the p110 ABD. In addition, p85α and p85β have a Rho GTPase activating protein (GAP) region and N-terminal portions containing an SH3 region. The SH2 regions in p85 recognize the same phosphotyrosine motif and allow the enzyme to associate with activated tyrosine kinase receptors (RTKs) and other intracellular phosphotyrosine containing proteins
[13][20].
Class IA enzymes are stimulated by cytokines, receptor tyrosine kinases (RTKs), G protein coupled receptors (GPCRs), and the small G protein Ras
[33]. These receptors recognize interleukin-6 (IL-6), hormones, epidermal growth factors (EGF), insulin-like growth factors (IGF), and inflammatory stimuli such as LPS, and the cellular signals known to be modulated comprise growth, survival, cell proliferation, and inflammatory processes
[17][34].
Class IB enzymes are dimers of the p110γ polypeptide complexed with an adaptor subunit, p101 or p84 and p87
[28][35]. Subunit p101 or p84 bind to a single p110γ polypeptide to obtain two heterodimers, p101/p110γ or p84/p110γ, both known as “PI3Kγ”
[14][29][36]. PI3Kγ is expressed in both tissues and cells, with the highest levels of distribution in myeloid cells
[37][38]. Differently from class IA catalytic subunits, p110γ contains a Gβγ binding domain in the catalytic region at the C-terminal, and it is mainly stimulated by GPCRs, thanks to the interaction of its the regulatory subunit with the subunit of trimeric G protein and therefore distinct from class IA PI3Ks, which instead are activated downstream of RTKs
[39].
Each of the Class I PI3K isoforms catalyzes the phosphorylation of PtdIns (4,5)P2 to generate PtdIns (3,4,5)P3, known as “PIP3”, and the substantial difference between them appears to be in their adaptation to upstream regulation by receptor transduction pathways
[14][29][36].
Class II PI3Ks have only one catalytic protein present in three different isoforms (PI3K-C2α, PI3K-C2β, and PI3K-C2γ), which preferentially uses PtdIns or PtdIns4P as substrates at the plasma membrane or endosomes, therefore forming both PtdIns3P and PtdIns (3,4)P2
[30][40]. These enzymes lack the regulatory-subunit binding region, but possess a Ras binding region and the PI3K core. Distinctively, they all possess a C-terminus extension consisting of a Phox homology (PX) region, a second C2 region, and several additional protein regions at their N-terminal region
[30].
Many studies show that several stimuli can activate these isoforms, including hormones
[30][41], chemokines such as monocyte chemotactic protein 1 (MCP1), cytokines such as tumor necrosis factor α (TNFα)
[42], growth factors such as EGF and stem cell factor (SCF)
[43], integrins
[44], and phospholipids such as lysophosphatidic acid (LPA)
[45].
These studies show that only some class II PI3K isoforms can be triggered downstream of RTKs
[30][43] and GPCRs
[45]. The cellular activity of Class II PI3K remains poorly characterized with respect to class I kinases. Furthermore, the substrate used by these enzymes is PtdIns4P
[30].
Lastly, class III PI3K consists of a single catalytic subunit termed Vps34 (vacuolar protein sorting 34), and its activity is modulated by the Vps15 kinase (known as p150 in mammalian cells) with which it binds
[46]. Vps34 phosphorylates PtdIns to form PtdIns3P with certain intracellular structures such as endosomes and, through binding to distinct effector regions, modulates the activity and fate of these vesicular structures
[47].
Vps34 is a lipid kinase that mediates cell signaling through mammalian target of rapamycin (mTOR), indicating a possible role in modulating cell growth
[47].
2.2. PI3K Signaling Pathway Activation
The typical activation manner of one or more isoforms of PI3Ks enzymes begins with the binding of a ligand to RTKs or GPCRs through a regulatory subunit, such as p85. In basal conditions, p85 associates with the N-terminal of the catalytic p110 subunit through its iSH2 region, inhibiting its function. Following appropriate cellular stimuli as many cytokines and growth factors, as well as by insulin and LPS
[14], the SH2 regions bind to activated receptors or adapter proteins, and this phosphotyrosine binding leads to allosteric activation of the p110 catalytic subunits of PI3K
[33][48]. In particular, the activation of class I PI3Ks converts the cellular membrane phospholipid PtdIns (4,5)P2 to PIP3
[14][48], whereas PtdIns (3,4)P2 can originate from PIP3 through the SHIP family of phosphatases (SHIP-1 and SHIP-2)
[18][49][50]. In addition, the class II PI3Ks using PI4P as a substrate can also produce PI3,4P2
[17]. Although the activation of specific PI3K isoform in a given cellular process can be different, the ultimate result is the same: repositioning and activity of numerous signaling proteins through binding to conserved regions like the PH (pleckstrin-homology) regions.
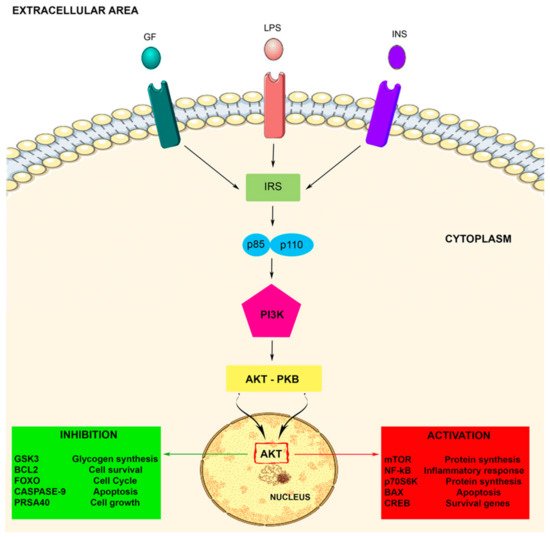
Therefore, PIP3 or PI3,4P2 as second messengers function as ligands to call back the PH region containing proteins at the inner side of the plasma membrane. These proteins including Akt (protein serine/threonine kinase) (also termed PKB) and PDK1 (phosphoinositide dependent kinase 1), which share a PH region selective for PIP3 and/or PtdIns 3,4-P2
[29]. Akt consists of three isoforms (Akt1, Akt2, and Akt3), which share a similar architecture: an N-terminus PH region, a central serine/threonine catalytic region, and a small C-terminus regulatory region. The PH region is critical for the PI3K/Akt signaling transduction
[51].
Akt activation allows the movement of the protein kinase towards the cytoplasm and nucleus, where it modulates numerous downstream proteins such as the Bcl-2 antagonist of cell death, caspase 9, glycogen synthase kinase-3 beta (GSK3β), p70 S6 kinase (p70S6K), fork head transcription factors (FOXOs), proline rich Akt substrate of 40kDa (PRAS40), nuclear factor-kappa B (NF-kB), and mTOR, either positively or negatively. These target proteins are implicated in several cell biological activities such as protein synthesis, cell proliferation, cell motility, metabolism, survival, apoptosis, cell growth, and neuroinflammatory disorders
[33][48][51]. Therefore, Akt represents a major mediator of PI3K signals, and the PI3K/Akt signaling pathway plays important roles in the cellular responses (see ).
Figure 1. The PI3K-Akt signaling pathway. The PI3K-Akt pathway is involved in some crucial cellular processes including protein synthesis and cell proliferation and survival. The PI3K/Akt pathway is activated by factors that initiate the PI3K signaling pathway via intermediate molecules (IRS), playing an important regulatory role in many cellular survival pathways. The pathway can be activated by a variety of signals, including growth factors (GF), LPS, and insulin (INS), targeting several downstream molecules. This activation is able to modulate cell activities, including cell proliferation, glucose metabolism, cell survival, cell cycle, protein synthesis, and neuronal morphology and plasticity.
3. Role of PI3K in Microglia Activity
It has been well established that neuroinflammation is an intricate inflammatory reaction that may occur within the peripheral nervous system or in the CNS, being generally triggered by tissue injury and infectious agents. It is an extremely dynamic and complex adaptive process, which definitely has the purpose of containing cellular damage, maintaining brain tissue integrity, as well as restoring tissue homeostasis. The production of pro-inflammatory cytokines in glial cells, especially in microglia and astrocytes, represents an intersection point between the immune and nervous systems, assigning to microglia the role of the predominant supplier of inflammatory mediators in the CNS
[52].
Microglia, astrocytes, and neurons, as well as all the CNS resident cells actively participate in neuroinflammation: among all, microglia and astrocytes seem to contribute extensively, playing a pivotal role in neuroinflammatory response regulation
[53].
Microglia and astrocytes are therefore actually believed to be responsible for the innate immune response in CNS, expressing a defense function against pathogens and damage. In this regard, even if reactive astrocytes seem able to coincide with neuroinflammation, after activation of microglia and neural injury, microglia remain, without any doubt, the most important component of neuroinflammation
[54].
In this regard, it is well understood that microglia scan the microenvironment continuously by generating factors that influence astrocytes and neuron that are in close proximity, triggering an inflammatory response that further includes a temporary, self-limiting response through the immune system and initiates tissue repair. During pathological conditions, when normal resolution mechanisms fail, an irregular activation and development of inflammatory factors results in a chronic neuroinflammatory state
[55].
It has been shown that microglia can either activate neuroinflammatory pathways that can lead to a progressive neurodegeneration or alternatively can promote neuroprotection by downregulation of inflammation and stimulation of neuronal repair. Therefore, microglia should not be considered as a uniform entity, but rather as a heterogeneous community with sub-populations that can be recognized on the basis of their different abilities to operate and perform various functions
[56].
In particular, a microglial polarization process is well recognized, suggesting the existence of two different forms of microglial cell response, one associated with neuroinflammation, while the other associated with neuroprotection. Moreover, it seems that microglial cells polarization process involves the microglial receptors leading to cellular signal transduction through a variety of intracellular pathways, such as MAPKs, STATs, PI3K, and NF-κB. However, to date, knowledge about microglial cell polarization during neuroinflammation is still limited, and it is the result of a restrained number of recent studies concerning stroke, depression, neuroinflammation, or PD
[34].
In this context, recent studies have focused substantial attention on the intracellular pathway that involves PI3K and the Akt kinase (PI3K/Akt). In addition, an important role of the PI3K/Akt cascade in the functions of microglial cells was recently reported. The activation of this signaling cascade is initiated by many cytokines and growth factors, as well as LPS, suggesting that the PI3K/Akt signaling pathway achieved via its specific receptor is crucial in LPS induced inflammation, playing a significant role in the microglial activation pathway, as well as in neuroinflammation
[15]. As a consequence, regulation of the PI3K/Akt axis is to be considered a possible approach in order to develop new treatments for neuropathological disorders by the inhibition of proinflammatory gene expression.
In line with this is the observation that curcumin seems to be able attenuate the release of the proinflammatory mediators in BV-2 microglia stimulated with LPS. This may be possible thanks to the suppression of the NF-κB pathway, obtained by the PI3K/Akt signaling pathway downregulation, which could cause a restriction of LPS induced inflammatory responses
[57].
It seems that in CNS, the PI3K/Akt pathway can represent an important signaling for neuroprotection. Nevertheless, some possible cytodynamics involving PI3K/Akt and its relation to progressive neurodegenerative disease remain an area of intense interest for research on neuroinflammation and related diseases. Moreover, it has become increasingly evident that several active biomolecules seem able to target the impaired PI3K/Akt balance and, for this reason, could significantly contribute to neuroprotection. This is extremely important in terms of longevity and could represent a promising target for therapeutical strategies improvement
[58].
 +1 credit
+1 credit