Immune activation in the central nervous system involves mostly microglia in response to pathogen invasion or tissue damage, which react, promoting a self-limiting inflammatory response aimed to restore homeostasis. However, prolonged, uncontrolled inflammation may result in the production by microglia of neurotoxic factors that lead to the amplification of the disease state and tissue damage. In particular, specific inducers of inflammation associated with neurodegenerative diseases activate inflammatory processes that result in the production of a number of mediators and cytokines that enhance neurodegenerative processes. Phosphoinositide 3-kinases (PI3Ks) constitute a family of enzymes regulating a wide range of activity, including signal transduction.
- neurodegeneration
- inflammation
- PI3K
- microglia
- signaling pathway
1. Introduction
2. PI3K Signaling Pathway
2.1. Classes of PI3K Enzymes
| PI3k | Expression | Ref |
|---|---|---|
| p110 α | Ubiquitous | [14][23] |
| p110 β | Ubiquitous | [14][23][24] |
| p110 δ | Immune cells, Neurons and Microglia, Spleen, Platelets, Endothelial Cells | [25] |
| p85 α | Ubiquitous | [24] |
| p55 α | Brain, Muscle | [26] |
| p50 α | Liver, Kidney, Brain, T Cells | [25] |
| p85 β | Ubiquitous | [24][26] |
| p55 γ | Brain, Testis, Liver, Muscle, Fat, Spleen | [24] |
| p110 γ | Immune Cells, Heart, Pancreas, Liver, Skeletal Muscle | [25][27] |
| p101 | Immune Cells, Mast Cells | [26][28][29] |
| p84/p87 | Immune cells, Mast Cell, Heart | [26][28][29] |
| pI3k-C2 | Ubiquitous | [30] |
| pI3k-C2β | Ubiquitous | [30] |
| pI3k-C2 γ | Liver, Prostate, Breast, Salivary Gland | [31] |
| Vps34 | Ubiquitous | [32] |
| Vps15 | Ubiquitous | [26] |
2.2. PI3K Signaling Pathway Activation
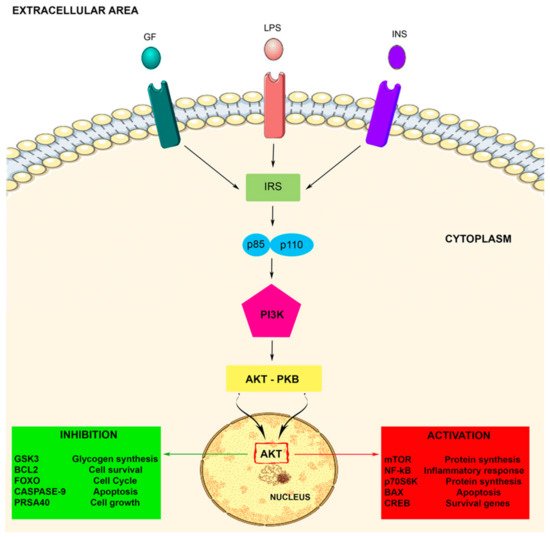
3. Role of PI3K in Microglia Activity
This entry is adapted from the peer-reviewed paper 10.3390/biom10010137
References
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell. Biol. 2000, 1, 120–129.
- Hoglund, K.; Salter, H. Molecular biomarkers of neurodegeneration. Expert Rev. Mol. Diagn. 2013, 13, 845–861.
- Frank-Cannon, T.C.; Alto, L.T.; McAlpine, F.E.; Tansey, M.G. Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 2009, 4, 47.
- Heneka, M.T.; Kummer, M.P.; Latz, E. Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol. 2014, 14, 463–477.
- Amor, S.; Puentes, F.; Baker, D.; Van der Valk, P. Inflammation in neurodegenerative diseases. Immunology 2010, 129, 154–169.
- Gendelman, H. Neural immunity: Friend or foe? J. Neurovirol. 2002, 8, 474–479.
- McManus, R.M.; Heneka, M.T. Role of neuroinflammation in neurodegeneration: New insights. Alzheimer Res. Ther. 2017, 9, 14.
- Glass, C.K.; Saijo, K.; Winner, B.; Marchetto, M.C.; Gage, F.H. Mechanisms underlying inflammation in neurodegeneration. Cell 2010, 140, 918–934.
- Lehnardt, S. Innate immunity and neuroinflammation in the CNS: The role of microglia in Toll-like R in cancer. Nat. Rev. Drug Discov. 2010, 8, 627–644.
- Holtman, I.R.; Raj, D.D.; Miller, J.A.; Schaafsma, W.; Yin, Z.; Brouwer, N.; Wes, P.D.; Möller, T.; Orre, M.; Kamphuis, W.; et al. Induction of a common microglia gene expression signature by aging and neurodegenerative conditions: A co-expression meta-analysis. Acta Neuropathol. Commun. 2015, 3, 31.
- Keren-Shaul, H.; Spinrad, A.; Weiner, A.; Matcovitch-Natan, O.; Dvir-Szternfeld, R.; Ulland, T.K.; David, E.; Baruch, K.; Lara-Astaiso, D.; Toth, B.; et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 2015, 169, 1276–1290.
- Orre, M.; Kamphuis, W.; Osborn, L.M.; Jansen, A.H.P.; Kooijman, L.; Bossers, K.; Hol, E.M. Isolation of glia from Alzheimer’s mice reveals inflammation and dysfunction. Neurobiol. Aging. 2014, 35, 2746–2760.
- Vanhaesebroeck, B.; Whitehead, M.A.; Piñeiro, R. Molecules in medicine mini-review: Isoforms of PI3K in biology and disease. J. Mol. Med. (Berl) 2016, 94, 5–11.
- Vanhaesebroeck, B.; Guillermet-Guibert, J.; Graupera, M.; Bilanges, B. The emerging mechanisms of isoform-specific PI3K signalling. Nat. Rev. Mol. Cell. Biol. 2010, 11, 329–341.
- Saponaro, C.; Cianciulli, A.; Calvello, R.; Dragone, T.; Iacobazzi, F.; Panaro, M.A. The PI3K/Akt pathway is required for LPS activation of microglial cells. Immunopharmacol. Immunotoxicol. 2012, 34, 858–865.
- Sun, N.; Wang, H.; Ma, L.; Lei, P.; Zhang, Q. Ghrelin attenuates brain injury in septic mice via PI3K/Akt signalling activation. Brain. Res. Bull. 2016, 124, 278–285.
- Hawkins, P.T.; Stephens, L.R. Emerging evidence of signalling roles for PI(3,4)P2 in Class I and II PI3K-regulated pathways. Biochem. Soc. Trans. 2016, 44, 307–314.
- Deane, J.A.; Fruman, D.A. Phosphoinositide 3-kinase: Diverse roles in immune cell activation. Annu. Rev. Immunol. 2004, 22, 563–598.
- Kitagishi, Y.; Kobayashi, M.; Kikuta, K.; Matsuda, S. Roles of PI3K/AKT/GSK3/mTOR Pathway in Cell Signalling of Mental Illnesses. Depress. Res. Treat. 2012, 2012, 752563.
- Amzel, L.M.; Huang, C.H.; Mandelker, D.; Lengauer, C.; Gabelli, S.B.; Vogelstein, B. Structural comparisons of class I phosphoinositide 3-kinases. Nat. Rev. Cancer 2008, 8, 665–669.
- Fruman, D.A.; Bismuth, G. Fine tuning the immune response with PI3K. Immunol. Rev. 2009, 228, 253–272.
- Whitehead, M.A.; Bombardieri, M.; Pitzalis, C.; Vanhaesebroeck, B. Isoform-selective induction of human p110δ PI3K expression by TNFα: Identification of a new and inducible PIK3CD promoter. Biochem. J. 2012, 443, 857–867.
- Leinders, M.; Koehrn, F.J.; Bartok, B.; Boyle, D.L.; Shubayev, V.; Kalcheva, I.; Yu, N.K.; Park, J.; Kaang, B.K.; Hefferan, M.P.; et al. Differential distribution of PI3K isoforms in spinal cord and dorsal root ganglia: Potential roles in acute inflammatory pain. Pain 2014, 155, 1150–1160.
- Geering, B.; Cutillas, P.R.; Nock, G.; Gharbi, S.I.; Vanhaesebroeck, B. Class IA phosphoinositide 3-kinases are obligate p85-p110 heterodimers. Proc. Natl. Acad. Sci. USA 2007, 104, 7809–7814.
- Fruman, D.A.; Chiu, H.; Hopkins, B.D.; Bagrodia, S.; Cantley, L.C.; Abraham, R.T. The PI3K Pathway in Human Disease. Cell 2017, 170, 605–635.
- Kok, K.; Geering, B.; Vanhaesebroeck, B. Regulation of phosphoinositide 3-kinase expression in health and disease. Trends. Biochem. Sci. 2009, 34, 115–127.
- Zhou, Z.; Tang, M.; Liu, Y.; Zhang, Z.; Lu, R.; Lu, J. Apigenin inhibits cell proliferation, migration, and invasion by targeting Akt in the A549 human lung cancer cell line. Anticancer Drugs 2017, 28, 446–456.
- Suire, S.; Coadwell, J.; Ferguson, G.J.; Davidson, K.; Hawkins, P.; Stephens, L. p84, a new G beta gamma-activated regulatory subunit of the type IB phosphoinositide 3-kinase p110 gamma. Curr. Biol. 2005, 15, 566–570.
- Hawkins, P.T.; Stephens, L.R. PI3K signalling in inflammation. Biochim. Biophys. Acta. 2015, 1851, 882–897.
- Falasca, M.; Maffucci, T. Regulation and cellular functions of class II phosphoinositide 3-kinases. Biochem. J. 2012, 443, 587–601.
- Hennessy, B.T.; Smith, D.L.; Ram, P.T.; Lu, Y.; Mills, G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug. Discov. 2005, 4, 988–1004.
- Valet, C.; Severin, S.; Chicanne, G.; Laurent, P.A.; Gaits-Iacovoni, F.; Gratacap, M.P.; Payrastre, B. The role of class I, II and III PI 3-kinases in platelet production and activation and their implication in thrombosis. Adv. Biol. Regul. 2016, 61, 33–41.
- Zhu, J.; Wang, M.; Cao, B.; Hou, T.; Mao, X. Targeting the phosphatidylinositol 3-kinase/AKT pathway for the treatment of multiple myeloma. Curr. Med. Chem. 2014, 21, 3173–3187.
- Popiolek-Barczyk, K.; Mika, J. Targeting the Microglial Signalling Pathways: New Insights in the Modulation of Neuropathic Pain. Curr. Med. Chem. 2016, 23, 2908–2928.
- Voigt, P.; Dorner, M.B.; Schaefer, M. Characterization of p87(PIKAP), a novel regulatory subunit of phosphoinositide 3-kinase gamma that is highly expressed in heart and interacts with PDE3B. J. Biol. Chem. 2006, 281, 9977–9986.
- Fritsch, R.; Downward, J. SnapShot: Class I PI3K isoform signalling. Cell 2013, 154, 940–940.e1.
- Sasaki, T.; Irie-Sasaki, J.; Jones, R.G.; Oliveira-dos-Santos, A.J.; Stanford, W.L.; Bolon, B.; Wakeham, A.; Itie, A.; Bouchard, D.; Kozieradzki, I.; et al. Function of PI3Kγ in thymocyte development, T cell activation, and neutrophil migration. Science 2000, 287, 1040–1046.
- Costa, C.; Martin-Conte, E.L.; Hirsch, E. Phosphoinositide 3-kinase p110gamma in immunity. IUBMB Life 2011, 63, 707–771.
- Katso, R.; Okkenhaug, K.; Ahmadi, K.; White, S.; Timms, J.; Waterfield, M.D. Cellular function of phosphoinositide 3-kinases: Implications for development, immunity, homeostasis, and cancer. Annu. Rev. Cell. Dev. Biol. 2001, 17, 615–675.
- Posor, Y.; Eichhorn-Gruenig, M.; Puchkov, D.; Schöneberg, J.; Ullrich, A.; Lampe, A.; Müller, R.; Zarbakhsh, S.; Gulluni, F.; Hirsch, E.; et al. Spatiotemporal control of endocytosis by phosphatidylinositol-3,4-bisphosphate. Nature 2013, 499, 233–237.
- Brown, R.A.; Domin, J.; Arcaro, A.; Waterfield, M.D.; Shepherd, P.R. Insulin activates the α isoform of class II phosphoinositide 3-kinase. J. Biol. Chem. 1999, 274, 14529–14532.
- Ktori, C.; Shepherd, P.R.; O’Rourke, L. TNF-alpha and leptin activate the a-isoform of class II phosphoinositide 3-kinase. Biochem. Biophys. Res. Commun. 2003, 306, 139–143.
- Arcaro, A.; Khanzada, U.K.; Vanhaesebroeck, B.; Tetley, T.D.; Waterfield, M.D.; Seckl, M.J. Two distinct phosphoinositide 3-kinases mediate polypeptide growth factor-stimulated PKB activation. Embo J. 2002, 21, 5097–5108.
- Liu, P.; Cheng, H.; Roberts, T.M.; Zhao, J.J. Targeting the phosphoinositide 3-kinase pathway in cancer. Nat. Rev. Drug. Discov. 2009, 8, 627–644.
- Visnjić, D.; Curić, J.; Crljen, V.; Batinić, D.; Volinia, S.; Banfić, H. Nuclear phosphoinositide 3-kinase C2beta activation during G2/M phase of the cell cycle in HL-60 cells. Biochim. Biophys. Acta. 2003, 631, 61–71.
- Kim, D.I.; Lee, K.H.; Gabr, A.A.; Choi, G.E.; Kim, J.S.; Ko, S.H.; Han, H.J. Aβ-Induced Drp1 phosphorylation through Akt activation promotes excessive mitochondrial fission leading to neuronal apoptosis. Biochim. Biophys. Acta. 2016, 11, 2820–2834.
- Backer, J.M. The regulation and function of Class III PI3Ks: Novel roles for Vps34. Biochem. J. 2008, 410, 1–17.
- Hemmings, B.A.; Restuccia, D.F. PI3K-PKB/Akt pathway. Cold Spring Harb. Perspect. Biol. 2012, 4, a011189.
- Hamilton, M.J.; Ho, V.W.; Kuroda, E.; Ruschmann, J.; Antignano, F.; Lam, V.; Krystal, G. Role of SHIP in cancer. Exp. Hemat. 2011, 39, 2–13.
- Blero, D.; Payrastre, B.; Schurmans, S.; Erneux, C. Phosphoinositide phosphatases in a network of signalling reactions. Pflugers. Archiv. 2007, 455, 31–44.
- Manning, B.D.; Toker, A. AKT/PKB signalling: Navigating the network. Cell 2017, 169, 281–405.
- Tian, L.; Ma, L.; Kaarela, T.; Li, Z. Neuroimmune crosstalk in the central nervous system and its significance for neurological diseases. J. Neuroinflammation 2012, 9, 155.
- Shabab, T.; Khanabdali, R.; Moghadamtousi, S.Z.; Kadir, H.A.; Mohan, G. Neuroinflammation pathways: A general review. Int. J. Neurosci. 2017, 127, 624–633.
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967.
- Sochocka, M.; Diniz, B.S.; Leszek, J. Inflammatory Response in the CNS: Friend or Foe? Mol. Neurobiol. 2017, 54, 8071–8089.
- Gertig, U.; Hanisch, U.K. Microglial diversity by responses and responders. Front. Cell. Neurosci. 2014, 8, 101.
- Cianciulli, A.; Calvello, R.; Porro, C.; Trotta, T.; Salvatore, R.; Panaro, M.A. PI3k/Akt signalling pathway plays a crucial role in the anti-inflammatory effects of curcumin in LPS-activated microglia. Int. Immunopharmacol. 2016, 36, 282–290.
- Elstner, M.; Morris, C.M.; Heim, K.; Bender, A.; Mehta, D.; Jaros, E.; Klopstock, T.; Meitinger, T.; Turnbull, D.M.; Prokisch, H. Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol. 2011, 122, 75–86.