 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Candice M. Brown | + 5228 word(s) | 5228 | 2021-05-26 10:35:45 | | | |
| 2 | Conner Chen | Meta information modification | 5228 | 2021-06-10 12:42:46 | | |
Video Upload Options
Tissue-nonspecific alkaline phosphatase (TNAP) is an ectoenzyme bound to the plasma membranes of numerous cells via a glycosylphosphatidylinositol (GPI) moiety. TNAP is one of many proteins localized to Brain microvascular endothelial cells (BMECs), and is highly abundant in human and rodent cerebral microvessels [33]. There are four alkaline phosphatase (AP) isoenzymes in humans and they include: TNAP, germ cell alkaline phosphatase (GCAP), intestinal alkaline phosphatase (IAP), and placental alkaline phosphatase (PLAP). Although TNAP is ubiquitous in many tissue, it is most highly expressed in bone, liver, intestine, kidney, and brain, while the three other AP isoenzymes are expressed in the tissues for which they are named. TNAP is also highly expressed in cerebral microvessels.
1. TNAP Biology
Most of the current knowledge in TNAP biology has been generated from studies on its role in bone and mineral metabolism. This section summarizes the cell biology and gene expression of TNAP, with an emphasis on findings in endothelial cells (ECs) and BMECs.
1.1. Cell Biology
TNAP is bound to the plasma membranes of cells via a glycosylphosphatidylinositol (GPI) moiety and functions as an ectoenzyme [1][2][3]. Known substrates for TNAP under healthy conditions include inorganic pyrophosphates (PPi), vitamin B6, adenosine triphosphate (ATP), adenosine diphosphate (ADP), adenosine monophosphate (AMP), and phosphoethanolamine (PEA) [4][5]. For a more in-depth discussion on the structure and substrates of TNAP, the reader is referred to this excellent review [6]. TNAP’s role in bone and mineral metabolism is well-established based on numerous studies showing that diminished TNAP activity causes clinical hypophosphatasia (HPP) in both humans and rodents [7][8][9]. TNAP is also important for appropriate skeletal mineralization. Deficits in TNAP activity result in the accumulation of PPi and osteopontin (OPN), both of which suppress hydroxyapatite crystal growth and formation, i.e., a key inorganic constituent required for bone mineralization [10][11][12]. Furthermore, patients with a clinical hypophosphatasia diagnosis also present with seizures [13]. Subsequent studies have revealed that these seizures are the result of failure to transport vitamin B6 across the BBB. TNAP’s inability to hydrolyze vitamin B6 in the periphery has also been shown to decrease the availability of pyridoxal, a hydrolyzed form of vitamin B6, which is able to cross the BBB. Thus, diminished TNAP activity creates a deficiency of vitamin B6 in the brain, which, in turn, disrupts the synthesis of gamma aminobutyric acid (GABA), an important inhibitory neurotransmitter [14][15]. Despite numerous studies which have demonstrated presumed functions for TNAP in both brain and peripheral tissues, its role in cerebral microvessels remains vastly unexplored.
1.2. Global Gene Expression and Enzyme Activity
Although TNAP protein is present in all mammals and in most tissues, its gene (ALPL in humans, Alpl or Akp2 in mice) expression varies among different tissues [16]. In the first of its 12 exons, the 5′-untranslated region (UTR) contains either exon 1A or 1B by alternative transcription initiation [17]. Exon 1A is preferentially driven in osteoblasts, while exon 1B is more often initiated by the liver and kidney [18][19][20]. Using marmosets, the TNAP isoform found in brain has been shown to use the same promotor as bone; however, exon 1B is preferentially transcribed by mouse neurons [16]. TNAP activity is also important during mouse embryo development and appears to be the predominant AP seen in 7- to 14-day-old embryos and primordial germ cells [21]. As of May 2021, there are 411 mutations reported in the ALPL gene mutations database (http://alplmutationdatabase.hypophosphatasie.com/) (Access on: 13 May 2021), the majority of which are pathogenic loss-of-function mutations that result in HPP. In addition to deficits in bone and teeth development, the depleted TNAP activity in HPP also results in impaired neuronal function that manifests most commonly as seizures [9][13][22]. Conversely, abnormally high levels of TNAP activity in neurons can also result in seizures and neurological dysfunction [22][23][24]. This paradox presents an interesting dynamic where TNAP activity levels must be sustained within a specific range in order to maintain homeostasis within the nervous system.
1.3. Gene Expression and Enzyme Activity in BMECs
Two different TNAP isoforms, commonly described as bone TNAP and liver TNAP, are expressed in the brain. The bone-type TNAP transcript is expressed in human and mouse BMECs [16]. During development, TNAP activity in murine cerebral microvessels is delayed and not seen until postnatal day 10. In comparison, TNAP activity has been detected as early as embryonic day 15 in rats [16] and at 28 weeks of gestation in humans [25]. In addition, TNAP enzymatic activity in the brain parenchyma compared to cerebral microvessels differs from one species to another. For example, TNAP activity is weaker in the brain parenchyma compared to cerebral microvessels in humans, monkeys, rodents, guinea pigs, and cats [26][27][28][29]. Conversely, TNAP activity in cerebral microvessels is stronger in the brain parenchyma and weaker in rabbits, frogs, and chickens [26][30] as shown in Table 1. Furthermore, there are significant differences between TNAP activity in cerebral microvessels compared to peripheral microvessels. A comparative study assessing the TNAP activity in BMECs compared to peripheral endothelial cells from the aorta showed that TNAP activity in BMECs is highly elevated compared to peripheral endothelial cells [31]. In addition, Vorbrodt et al. showed that TNAP in the endothelial cells of the liver sinusoids was absent, while skeletal endothelial cells revealed a strong TNAP activity that was discontinuous or irregularly scattered across the plasma membrane compared to BMECs, which showed a continuous and uniform layer of strong TNAP enzyme activity [32]. Given these observations, species-specific and developmental differences in TNAP protein expression and enzyme activity need to be taken into account when elucidating the function of BMEC TNAP in animal models prior to translating these findings to human BMEC TNAP function. Additionally, other cell types that comprise the BBB such as astrocyte end-feet processes and pericytes also exhibit moderate TNAP activity [33][34][35]. Co-culture of BMECs with mixed glial cells/co-cultures in vitro has been shown to drastically increase TNAP protein, activity, and mRNA expression in BMECs compared to solo-cultures [36][37][38]. In another study, Tio et al. used a conditioned medium from astrocytes to demonstrate that secreted products from astrocytes were also capable of inducing an increase in TNAP activity in endothelial cells [39].
Table 1. Species-specific TNAP activity in the CNS.
| Animal Models | TNAP Activity in Cerebral Microvessels | Gene Name | Time Period When First Detected in CNS | Ref. |
|---|---|---|---|---|
| Zebrafish | Activity is present in the brain parenchyma and vessels | alpl | 11 h post-fertiilzation (hpf) | [40] |
| Mouse | Strong activity in vessels and weaker activity in the brain parenchyma | Alpl or Akp2 | Embryonic day 7–14 | [21][26] |
| Rat | Strong activity in vessels and weaker activity in the brain parenchyma | Alpl or Akp2 | Embryonic day 15 | [16][26] |
| Guinea Pig | Strong activity in vessels and weaker activity in the brain parenchyma | Alpl | - | [26] |
| Frog | Activity is absent on vessels but present on inner arachnoid and perineurium | alpl | - | [30] |
| Chicken | Weak activity in vessels and strong activity in the brain parenchyma | ALPL | Day 2 of the incubation period | [26][41] |
| Rabbit | Activity is absent in vessels but strong in brain parenchyma | ALPL | - | [26] |
| Cat | Strong activity in vessels and weaker activity in the brain parenchyma | ALPL | - | [26] |
| Rhesus Monkey | Strong activity in vessels and laminar activity pattern in the brain parenchyma (i.e., some areas/layers have higher activity than others) | ALPL | - | [42] |
| Human | Strong activity in vessels and laminar activity pattern in the brain parenchyma (i.e., some areas/layers have higher activity than others) | ALPL | 28 weeks of gestation | [25][43] |
2. Models and Tools to Study TNAP
One of the many challenges to elucidating the role of TNAP in health and disease has been the creation of appropriate genetic and pharmacological tools. This section will address their advantages and disadvantages.
2.1. Mouse Models
The life expectancy of mice with a global genetic deletion of the Alpl gene (Alpl−/−) averages 3–5 weeks due to HPP and neurological deficits such as seizures [14][44]. The pathophysiology of HPP is described in a recent review that provides an excellent overview of mouse models where Alpl is either genetically or chemically depleted [45]. Due to the side-effects from the HPP phenotype, Alpl null mice are inadequate models to study molecular or cellular functions associated with TNAP beyond young adulthood. Alternatively, other researchers have employed adult heterozygous transgenic mice (Alpl+/−) to explore the consequences of reduced TNAP activity [46][47][48]. Injection of a recombinant mineral-targeted TNAP lentivirus into neonatal mice has been shown to increase AP levels up to 60 days and reduced craniosynostosis [49][50]. Due to the wide and variable expression of TNAP in various cell types, TNAP-expressing cells can also be targeted using the Cre-Lox system. For example, using mice with tamoxifen-inducible inactivation of transforming growth factor β (TGF-β) in TNAP-expressing cells (Tnapcre;Tgfßr2fl/fl), researchers have shown that TNAP mitigates TGF-ß-dependent cardiac and skeletal muscle fibrosis through inactivation of SMAD2/3 transcription factors [51].
The majority of research in this field has focused on overexpression of TNAP in peripheral endothelial cells to study vascular calcification. Cell-specific mouse models generated using Cre recombinase transgenic mice and manipulation of the TNAP gene are described in Table 2. Conditional TNAP overexpression has been achieved using an HprtALPL knock-in mouse, which is described in [52]. Briefly, this model contains a floxed “stop cassette” and human ALPL cDNA is inserted into the hypoxanthine phosphoribosyltransferase (Hprt) locus on the X chromosome. Cross-breeding of the HprtALPL with Cre-expressing mice results in deletion of the stop cassette and constitutive ALPL expression within the target cell type. Because of the nature of this X-linked system, both homozygous (HprtALPL/ALPL) and heterozygous (HprtALPL/−) females are considered to be over-expressors, however HprtALPL/− females typically show a milder phenotype. For example, female TagIn-Cre+/−;HprtALPL/− mice showed a much milder medial vascular calcification (MVC) phenotype compared to male hemizygous TagIn-Cre+/−, HprtALPL/Y mice; however, the effects in female TagIn-Cre+/−;HprtALPL/ALPL mice were not described [52].
Table 2. Cell-Specific In Vivo Mouse Models for TNAP.
| Mouse Model Type | Cre-Mediated Target Cell | Preclinical Outcomes | Ref. |
|---|---|---|---|
| TNAP OE | Vascular smooth muscle cells (Tagln-Cre or SM22-Cre) | Mice had increased AP enzyme activity, increased systolic blood pressure, and increased vascular calcification. Male TNAP-OE mice lifespan was shorter than WT controls, as all died by 5 months of age. | [52] |
| TNAP OE | Endothelial cells (Tie2-Cre) | Increased AP activity was localized to the luminal side of the aorta and vascular networks of heart, lung, kidney, liver, small intestine, and pancreas. No skeletal abnormalities were detected; however, in the heart, kidney, mesentery, pancreas, spleen, and lung parenchyma there were calcified lesions in adult mice. | [53] |
| TNAP OE with “wicked high cholesterol” (C57BL/6J-LdlrHlb301/J) | Endothelial cells (Tie2-Cre) | Mice displayed increased AP activity in endothelial cells and increased sub-endothelial calcification nodules in their coronary arteries, which recapitulates murine atherosclerosis. | [54][55] |
| TNAP OE | Endothelial cells (Cdh5-Cre) | Over-expression of TNAP on endothelial cells resulted in increased survival and decreased clinical severity post-sepsis compared to controls. Locomotor activity in the last 5 min of open field testing was also increased in VE-cOE mice compared to controls. | [56] |
| TNAP KO | Osteoblasts and odontoblasts (Col1a1-Cre); Early limb bud mesenchyme and a subset of craniofacial mesenchyme (Prx1-Cre) |
While both Cre recombinase drivers result in similar phenotypes with regards to skeletal defects in cortical and trabecular bone, Prx1-cKO mouse long bones appeared more severely affected. Both models resulted in increased osteoclast numbers on alveolar bone surfaces and reduced alveolar bone height. Both models resulted in cementum and periodontal ligament defects, consistent with periodontal disease. | [57] |
| TNAP KO | Endothelial cells (Cdh5-Cre) | Primary BMECs revealed decreased barrier integrity compared to controls, which was mitigated after treatment with fasudil. | [58] (results reported in a preprint) |
Key: OE = overexpression, WT = wild-type, KO = knock-out.
More recently, Alplfl/fl mice were developed; these mice possess a floxed Alpl allele, allowing for cell-specific conditional knock-down of TNAP when crossed with Cre recombinase transgenic mice [57]. Our laboratory has used the Alplfl/fl mouse to elucidate the function of TNAP in BMECs. In contrast to the conditional models described previously, we selected a specific Cdh5-Cre driver, which codes for VE-cadherin, rather than a Tie-2-Cre driver to interrogate TNAP function in these cells. We selected the Tg(Cdh5-cre)7MliaA strain based on published reports that this specific strain exhibited the highest levels of reporter gene expression in the brain compared to other strains of Cdh5-Cre mice [59][60]. By crossing the Cdh5-Cre driver mouse with the HprtALPL mouse described above, our lab also generated a model of TNAP over-expression on endothelial cells (i.e., VE-cOE mice). Using this model, we observed an increase in survival and decreased clinical severity scores at 48 h post-sepsis, indicating to us that TNAP enzyme activity on endothelial cells is important for post-sepsis recovery [56]. More recently, we generated a mouse model with conditional deletion of endothelial TNAP by crossing the Cdh5-Cre driver mouse with the Alplfl/fl mouse (i.e., VE-cKO mice). Using primary BMEC cultures from VE-cKO mice, we observed decreased barrier integrity compared to Alplfl/fl controls, further indicating the protective effects that TNAP activity offers to the BBB. These observations have been reported in a preprint [58].
2.2. TNAP Pharmacological Tools and Pharmaceuticals
There are several commercially available TNAP inhibitors currently on the market. L-homoarginine (hArg) and levamisole are two of the oldest inhibitors; however, these inhibitors lack selectivity for specific isoenzymes and offer weak binding to TNAP [61][62]. Recent studies have shown that although hArg had no effect on TNAP levels in TNAP-overexpressing mice, it did offer some protection from myocardial remodeling through mechanisms unrelated to TNAP inhibition [54]. Although levamisole has been used in several studies to help identify the role that TNAP plays in health and disease [63][37][64], it is a reversible inhibitor, so cells treated with this drug rapidly regain TNAP activity [65]. In order to identify small molecule inhibitors to target specific AP isoforms, Millan and colleagues employed high-throughput screening approaches and identified several small molecule aryl sulfonamides as potent and selective TNAP inhibitors [64]. The most potent inhibitor identified in this study was 2,5-dimethoxy-N-(quinoline-3-yl) benzenesulfonamide, or MLS-0038949. This molecule is commercially available for purchase and most commonly referred to as TNAP inhibitor (TNAPi); this molecule is also targeted primarily for in vitro studies (personal communication, Dr. Jose Luis Millan). Further screening for additional candidates with higher selectivity and potency identified a second inhibitor, SBI-425, which is more appropriate for in vivo applications [66]. SBI-425 has been used in several studies related to vascular calcification [57][61][67] and by our own lab to identify its applicability for studying TNAP’s role in the brain. In collaboration with the developers of TNAPi and SBI-425, we demonstrated that the SBI-425 inhibitor is unable to cross the BBB in healthy mice [56]. Since TNAP is localized to both the luminal and abluminal surfaces of brain endothelial cells [32], the inability of SBI-425 to cross the BBB in health may be useful in studying the effects of brain endothelial luminal TNAP activity.
To date, there are few pharmaceuticals available for exogenous treatment to restore AP activity. Enzyme replacement therapy using asfotase alfa (brand name: STRENSIQ) is the first and only, thus-far, FDA-approved medication for patients with perinatal-, infantile-, and juvenile-onset HPP [68][69]. This drug works through the cleavage of inorganic pyrophosphate (PPi) to phosphate (Pi) so that Pi may bind with calcium to form hydroxyapatite crystals needed for healthy bones [10][70][71]. Overall, this treatment has shown to help improve physical function and health-related quality of life in adults with pediatric-onset HPP [67]. Recombinant human TNAP (rhTNAP) purified from rabbit transgenic milk samples and administered to LPS-infected mice resulted in increased survival [72]. Clinical trials using bovine intestinal AP (IAP) have shown functional improvement in patients with sepsis and ulcerative colitis [70][71][73][74]. However, due to side-effects, a human recombinant AP (recAP), which encompasses the stability afforded from placental AP and catalytic viability of IAP, was created and has been promising in clinical trials [75][76]. In preclinical studies recAP did not affect pulmonary inflammation or endothelial and epithelial dysfunction in rats [77]. In a Phase II clinical trial, recAP failed to improve short-term kidney function in patients with sepsis-associated acute kidney injury (sepsis-AKI), although an exploratory finding showed lower mortality in patients who received recAP compared to those who received a placebo [78]. A Phase III clinical trial is now in progress in North America and Europe to test recAP in a larger group of patients with sepsis-AKI.
3. TNAP in Brain Microvascular Endothelial Health and Function
TNAP enzyme activity is highly elevated in the BMECs within cerebral microvessels compared to ECs in the peripheral vasculature. Due to this unique, yet poorly understood aspect of TNAP biology, histological detection of AP (i.e., TNAP) activity has been employed as a tool by neuroscientists and anatomists for nearly a century [63]. Please refer to Figure 2 legend for methodology used to quantify TNAP activity. For the remainder of this review, any discussion of TNAP in brain ECs will generally refer to TNAP in BMECs.
3.1. TNAP and the Heterogeneity of the Brain’s Vasculature
The brain’s vasculature is heterogenous and displays an arteriovenous hierarchy similar to peripheral vascular beds. Moreover, the expression of a single protein or molecule differs across the cerebral arteriovenous vascular bed. For example, a recent study demonstrated that the expression of von Willebrand Factor (VWF) is increased in venous BMECs compared to brain arterial and capillary BMECs, whereas Mfsd2a protein is highly abundant in capillary BMECs compared to brain arterial and venous BMECs [79]. The localization of TNAP activity in the brain endothelium has been explored in earlier studies. Vorbrodt et al. showed that adult mouse capillary and arterial BMECs exhibit strong TNAP activity, while venules showed a complete lack of TNAP activity. Importantly, despite brain arterioles and capillaries exhibiting strong TNAP activity, electron microscopy showed that capillary TNAP activity is present on the luminal surface, whereas, arterioles demonstrate TNAP activity on the luminal and abluminal surfaces of BMECs [32]. Intriguingly, a recent study utilizing single-cell RNA sequencing demonstrated that TNAP protein, referred to as Alpl in this study, is also present in the cerebral venous endothelium. Furthermore, this same study also demonstrated that TNAP protein is highly expressed in brain ECs compared to astrocytes, pericytes, neurons, microglia, and oligodendrocytes [80]. It is unclear whether this new finding contradicts the earlier findings from the Vorbrodt et al. study, which showed an absence of TNAP activity on the venous endothelium using electron microscopy, or whether it simply demonstrates that the protein is present on the venous endothelium but its activity is absent. Regional, or spatial, brain microvascular TNAP activity is also likely to be important. A recent preliminary study from our laboratory showed that TNAP enzymatic activity is higher in the striatum and cortex, compared to hippocampal cerebral microvessels; in TNAP protein localization and/or enzymatic activity throughout brain microvasculature, a mechanistic role for TNAP in the BMECs has also begun to emerge. These results have been reported in a preprint [58].
3.2. Potential Mechanism(s) of Action for TNAP in the Brain’s Vasculature
The molecular mechanisms underlying TNAP function in the brain have been studied more thoroughly in neurons compared to BMECs. For an in-depth discussion of TNAP function in neurons, the reader is directed towards these excellent reviews [15][81][82]. A study from Deracinois et al. was the first to show that inhibition of TNAP using a pan-AP inhibitor (levamisole) in bovine capillary endothelial cells (BCECs) increased cellular barrier permeability to Lucifer Yellow (PeLY) [36]. Barrier integrity can also be quantified using the xCELLigence real time cell analyzer (RTCA) instrument, which measures focal adhesion of cells. By utilizing this barrier assay, recent data from our laboratory demonstrated that pharmacological inhibition of TNAP by the previously described highly specific TNAP inhibitor, TNAPi (see Section 2.2), in a human BBB endothelial cell line (hCMEC/D3 cells), increased paracellular barrier permeability. We have also shown that primary BMECs (pBMECs) cultured from adult mice with a conditional deletion of endothelial TNAP (VE-cKO mice, see Table 2) display diminished paracellular barrier permeability. Next, we demonstrated that loss of TNAP induced cytoskeletal remodeling, as quantified by phalloidin and vimentin immunofluorescence, via the increased BMEC expression of rho-associated protein kinase (ROCK) 1 and 2 proteins by using in-cell westerns (ICW). Lastly, we showed that fasudil, a pan-ROCK inhibitor, mitigated the worsened paracellular barrier integrity seen in TNAPi-treated hCMEC/D3 cells or VE-cKO pBMECs. These collective preliminary results have been reported in a preprint [58]. Taken together, our results demonstrate a novel mechanism through which TNAP is able to maintain barrier integrity in brain endothelial cells (Figure 1) and may explain why drugs like fasudil have shown some therapeutic efficacy in preclinical models of sepsis and stroke [83][84][85][86]. However, there are some potential limitations to this proposed mechanism. For example, the Deracinois et al. study showed that BCECs co-cultured with mixed glial cells did not exhibit an increase permeability to PeLY following levamisole treatment [36]. Consequently, it is unclear how this new mechanistic pathway may operate in vivo in the presence of other cell types found in the BBB. Future studies will need to address such limitations in vivo to understand how cell-cell interactions within the BBB contribute to TNAP function in BMECs.
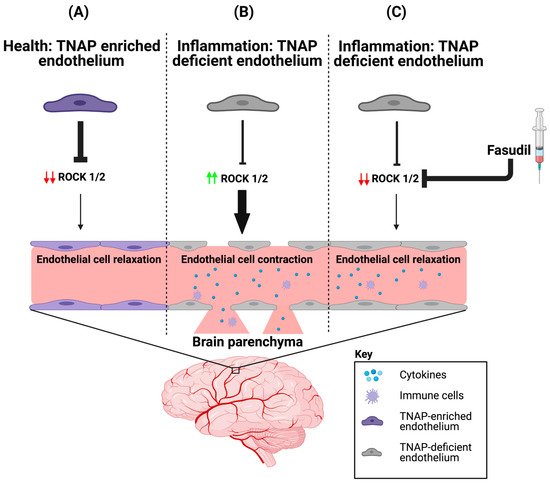
Figure 1. ROCK proteins are involved in BMEC TNAP function. (A) In health, BMECs are enriched with TNAP activity. We propose that the abundant TNAP activity decreases ROCK (1/2) protein expression, which maintains the endothelial cytoskeleton by ensuring that brain endothelial cells are in a relaxed state. (B) However during inflammation, BMEC TNAP activity decreases substantially, and this decrease promotes increased ROCK (1/2) protein expression (red arrows). Increased ROCK (1/2) protein expression (green arrows) leads to BMEC cytoskeletal contraction and ultimately alters paracellular barrier integrity. Disruption of the BMEC paracellular barrier allows for the infiltration of cytokines and immune cells into the brain parenchyma. (C) Therapeutic injection of fasudil (ROCK inhibitor) during inflammation decreases ROCK (1/2) protein expression (red arrows), thereby allowing for a maintenance of paracellular barrier integrity despite the loss of BMEC TNAP activity. BMEC: Brain microvascular endothelial cells; TNAP: Tissue-nonspecific alkaline phosphatase; ROCK: rho-associated protein kinase. Image credit: Biorender.
Several molecules such as retinol, cyclic AMP (cAMP), glucocorticoids, transforming growth factor-beta (TGF-β), interleukin-6 (IL-6), and basic fibroblast growth factor (bFGF) are known to modulate TNAP activity [63]. While retinol, cyclic cAMP, glucocorticoids, IL-6, and bFGF have been shown in previous studies to increase TNAP activity [81][82][87][88], TGF-β has been shown to suppress TNAP activity in brain ECs [88]. This non-exhaustive list of molecules highlights their potential importance in regulating TNAP function in brain ECs; however, the mechanisms through which these molecules modulate TNAP activity remain to be elucidated. Likewise, it remains unclear how the presence of TNAP activity on brain ECs influences targeted downstream pathways associated with these molecules.
4. A Role for Endothelial TNAP in the Brain’s Microvasculature in Aging and Disease
A significant number of studies have demonstrated a role for peripheral endothelial TNAP in disease [54][61][67][89]; however, there is a paucity of data on the function of brain microvascular TNAP activity in disease states. In this section, we examine emerging data for the role of brain microvascular TNAP in aging and disease.
4.1. Brain Microvascular TNAP in Disease
BBB dysfunction is a common neuropathological feature seen across many neuroinflammatory diseases [90]. Yet, there are no specific therapies that effectively mitigate BBB dysfunction to treat neurological disorders. Historically, quantification of TNAP activity has been used as a histological marker to detect changes in cerebral microvessel density or morphology in neurological disease. In spite of widespread TNAP activity as a histological tool, there was a minimal emphasis on physiological explanation regarding why the loss of TNAP activity occurred and how the loss of TNAP activity might affect disease progression [63]. This perspective is exemplified in an earlier study that quantified TNAP activity in cerebral microvessels to demonstrate that patients with leukoaraiosis (LA) exhibited decreased vascular density [91]. Yet, it was not evident from this study why TNAP activity was decreased in patients with LA compared to age-matched controls. It was also unclear if the loss of TNAP activity in LA was due to the loss of cerebral microvessels since the study did not utilize appropriate markers for the vasculature such as CD31, CD34, or collagen type IV [92]. Owing to these findings, our laboratory identified several important questions for future investigations: (1) Is the loss of TNAP activity on cerebral microvessels specific to a unitary disease state; (2) Does the loss of TNAP activity equate to the loss of cerebral microvessels; (3) Does diminished TNAP activity in cerebral microvessels result in alterations of BBB integrity; and (4) What are the relevant long-term neuroinflammatory and behavioral implications of the loss of TNAP activity in BMECs? Recent studies from our laboratory have begun to answer these new questions.
To elucidate whether BMEC TNAP activity was altered in systemic inflammation, we utilized the cecal ligation and puncture (CLP) model of sepsis to determine whether TNAP activity changed post-sepsis, and if early changes in TNAP activity were sustained long-term, i.e., seven days, into late sepsis. Our results showed that loss of TNAP activity is present as early as 24 h post-sepsis [93] and this loss was sustained up to seven days post-sepsis [94]. However, it was unclear whether the resultant loss of TNAP activity was due to cerebral microvessel loss as postulated in earlier studies [91]. Therefore, we combined CD31-immunolabelling of cerebral microvessels with histological detection of TNAP activity. Our results showed that the loss of TNAP activity was not due to a loss of cerebral microvessels, as the density of CD31+ cerebral microvessels remained consistent while TNAP activity decreased in the presence of septic injury. This important finding refutes the utility of brain microvascular TNAP activity as a measure of cerebral microvascular density since this activity changes with disease. More importantly, we showed that the loss of TNAP activity on cerebral microvessels coupled the loss of barrier integrity through the loss of junctional protein claudin-5. We also demonstrated that loss of TNAP activity was associated with increased microgliosis, astrocyte proliferation, and the infiltration of an endogenous permeability marker, immunoglobulin G (IgG), into the brain of septic mice [94]. To further demonstrate the role of brain microvascular TNAP in disease, we intraperitoneally injected septic mice with vehicle or SBI-425, an in vivo TNAP inhibitor discussed in Section 2.2, and subsequently examined claudin-5 expression. Our results showed that claudin-5 expression was decreased in SBI-425-treated septic mice compared to vehicle-treated septic mice. Since sepsis survivors are burdened with long-term cognitive impairments [95], we also examined whether the loss of TNAP in cerebral microvessels was coupled to behavioral deficits at seven days post-sepsis. Our results demonstrated that septic mice displayed an impaired sensorimotor phenotype but had no memory deficits. We speculate that the impairment in sensorimotor dysfunction most likely resulted from the observed loss of TNAP in the cortex, striatum, and spinal cord. In contrast, we attribute the lack of observed memory deficits to the absence of any changes between sham and septic mice when hippocampal TNAP activity was quantified [94]. Nevertheless, it remains unclear whether hippocampal TNAP activity is decreased at later timepoints (>seven days post-sepsis) when memory loss has been observed in other studies in septic mice [96][97].
Two important questions emerged after these intriguing findings: (1) What molecules initiated the loss of BMEC TNAP activity in sepsis, and (2) Was the loss of TNAP activity unique to sepsis or was it present in acute neuroinflammatory conditions with BBB disruption such as stroke? Indeed, our laboratory recently reported in a preprint that the loss of TNAP activity in sepsis manifests similarly in the cortical and striatal penumbra following 60 min transient middle cerebral artery occlusion, a model of experimental ischemic stroke, in mice [58]. Since animal models of sepsis and stroke have been shown to exhibit a rapid increase in systemic and cerebral pro-inflammatory cytokines [89][98][99][100], we hypothesized that the presence of pro-inflammatory cytokines would decrease BMEC TNAP activity. As expected, treatment of hCMEC/D3 cells with TNF-α and interferon-gamma (IFN-γ) significantly decreased BMEC TNAP activity, thereby resulting in both the loss of barrier integrity similarly to what was observed when BMECs were treated with TNAPi alone [58] (results reported in a preprint). Taken together, these novel findings have begun to reveal a functional understanding of brain microvascular TNAP function in acute disease conditions. Conversely, a comparable understanding brain microvascular TNAP’s function in chronic neuroinflammatory disease conditions such as AD remains to be elucidated. We present here for the first time preliminary evidence suggesting that brain microvascular TNAP activity is decreased in a model of AD (APPSwDI/Nos2−/− or CVN-AD [101]) compared to wild-type (WT) mice (Figure 2). The CVN-AD mouse exhibits AD-like pathologies and behavioral outcomes such as parenchymal beta-amyloid deposition, vascular amyloid angiopathy, neuronal loss, and tau hyperphosphorylation independent of a tau mutation. Additionally, the mouse also exhibits cognitive and sensorimotor deficits along with metabolic and sleep disturbances, all of which have been characterized in AD patients [102][103]. This result contrasts with earlier studies, which demonstrated that elevated neuronal TNAP activity exacerbates tau toxicity through the dephosphorylation and subsequent destabilization of extracellular tau released into the brain parenchyma from dying neurons [104]. Further studies are necessary to dissect the role(s) of both brain microvascular TNAP and neuronal TNAP in AD pathogenesis.
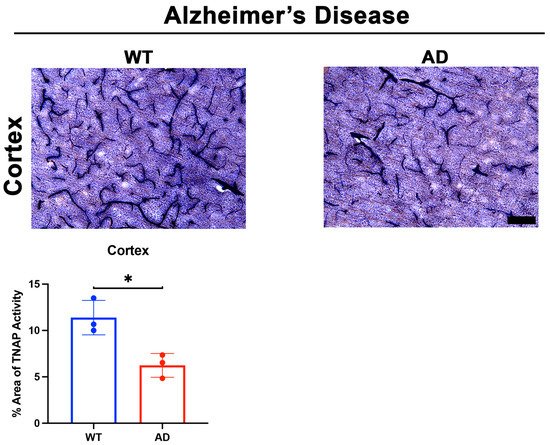
Figure 2. TNAP activity in a mouse model of AD. Preliminary data from male CVN-AD (AD) mice demonstrate a significant decrease (p = 0.01) in brain microvascular TNAP activity in the cortex compared to WT (C57BL/6J) mice. Data were analyzed using unpaired student’s t-test and n = 3/group. TNAP activity tissue histology was performed as previously described [94]. Briefly, tissue sections were evaluated for alkaline phosphatase activity using the BCIP/NBT substrate kit (SK-5400, Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions. BCIP/NBT is an AP substrate that undergoes an oxidation/reduction reaction when dephosphorylated by AP. The result of this dephosphorylation is visualized as a blue color change at the site of the reaction. This substrate can be used to measure AP activity in tissue sections or cells. Enzyme activity in sections is quantified using ImageJ. TNAP: Tissue-nonspecific alkaline phosphatase; AD: Alzheimer’s disease; WT: Wild-type. Images taken at 20× magnification and scale bar = 75 µm. * indicates p < 0.05.
4.2. Brain Microvascular TNAP in Aging
Aging increases susceptibility of brain ECs to injury or toxins [105]. Preclinical studies utilizing plasma substitution therapies from aged mice into young mice have elucidated the impact of aging on brain EC function and zonation of the cerebrovasculature [106][107]. While a clear role for BMEC TNAP in aging is not well- understood, recent findings suggest that brain microvascular TNAP protein, referred to as Alpl, is decreased in the hippocampus of aged mice (20 months old) compared to young mice (3 months) [107]. Given the observation of increased TNAP protein in aging, we examined whether brain microvascular TNAP activity is also increased with aging. As shown in Figure 3, we found that TNAP activity is increased in cerebral microvessels of the cortex, striatum, and hippocampus (CA3 shown) in 14-month-old mice compared to 4-month-old mice. These results suggest a dichotomy in brain microvascular TNAP function whereby TNAP activity is decreased in acute injuries like sepsis and ischemic stroke, while TNAP activity is increased in aging. The implications of the increased brain microvascular TNAP protein were further explored in a new report, which showed that intraperitoneal injection of TNAPi, i.e., MLS-0038949, in aged mice enhanced transcytosis of plasma proteins into the brain parenchyma [80]. To our knowledge, these are the only studies that have investigated a functional role for brain microvascular TNAP in aging. Importantly, transcriptomic profiling has shown that vessel segments, i.e., arteries, capillaries, or venules, exhibit different changes in their transcriptomic profiles during aging [107]. Overall, these findings emphasize the need to consider the impact of zonation, differential functions of the vascular tree, and as well as regional differences when studying BMEC TNAP function in normal health and in disease.
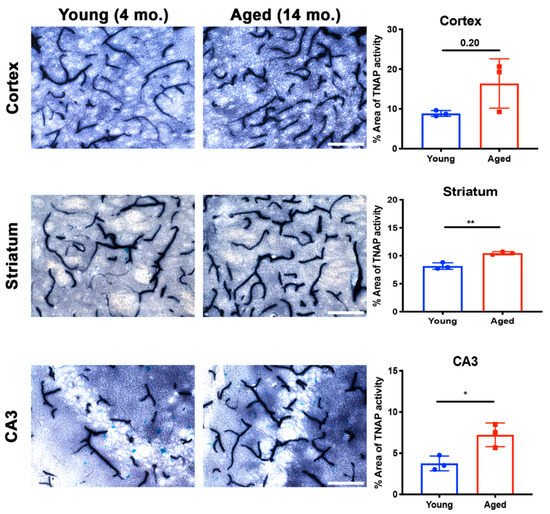
Figure 3. Brain microvascular TNAP activity in aging. The striatum (p = 0.004) and hippocampal CA3 (p = 0.02) regions revealed a significant increase in brain microvascular TNAP activity in aged male mice (14-month old) compared to young male mice (4-month old). The cortex also revealed an increase in TNAP activity in aged mice; however, this increase was not statistically significant (p = 0.20). Data were analyzed using unpaired Student’s t-test with n = 3/group. Histological staining to quantify TNAP activity was performed as previously described [94]; also see Figure 2 for a detailed description of this method. TNAP: Tissue-nonspecific alkaline phosphatase. Images taken at 40× magnification and scale bar = 100 µm. * indicates p < 0.05 and ** p < 0.01.
References
- Low, M.G. Biochemistry of the glycosyl-phosphatidylinositol membrane protein anchors. Biochem. J. 1987, 244, 1–13.
- Low, M.G.; Zilversmit, D.B. Role of phosphatidylinositol in attachment of alkaline phosphatase to membranes. Biochemistry 1980, 19, 3913–3918.
- Betz, A.L.; Firth, J.A.; Goldstein, G.W. Polarity of the blood-brain barrier: Distribution of enzymes between the luminal and antiluminal membranes of brain capillary endothelial cells. Brain Res. 1980, 192, 17–28.
- Whyte, M.P.; Landt, M.; Ryan, L.M.; Mulivor, R.A.; Henthorn, P.S.; Fedde, K.N.; Mahuren, J.D.; Coburn, S.P. Alkaline phosphatase: Placental and tissue-nonspecific isoenzymes hydrolyze phosphoethanolamine, inorganic pyrophosphate, and pyridoxal 5’-phosphate. Substrate accumulation in carriers of hypophosphatasia corrects during pregnancy. J. Clin. Investig. 1995, 95, 1440–1445.
- Say, J.; Ciuffi, K.; Furriel, R.P.; Ciancaglini, P.; Leone, F.A. Alkaline phosphatase from rat osseous plates: Purification and biochemical characterization of a soluble form. Biochim. Biophys. Acta BBA Gen. Subj. 1991, 1074, 256–262.
- Graser, S.; Liedtke, D.; Jakob, F. TNAP as a New Player in Chronic Inflammatory Conditions and Metabolism. Int. J. Mol. Sci. 2021, 22, 919.
- Moore, C.A.; Ward, J.C.; Rivas, M.L.; Magill, H.L.; Whyte, M.P. Infantile hypophosphatasia: Autosomal recessive transmission to two related sibships. Am. J. Med Genet. 1990, 36, 15–22.
- Barvencik, F.; Beil, F.T.; Gebauer, M.; Busse, B.; Koehne, T.; Seitz, S.; Zustin, J.; Pogoda, P.; Schinke, T.; Amling, M. Skeletal mineralization defects in adult hypophosphatasia—a clinical and histological analysis. Osteoporos. Int. 2011, 22, 2667–2675.
- Fedde, K.N.; Blair, L.; Silverstein, J.; Coburn, S.P.; Ryan, L.M.; Weinstein, R.S.; Waymire, K.; Narisawa, S.; Millan, J.L.; MacGregor, G.R.; et al. Alkaline Phosphatase Knock-Out Mice Recapitulate the Metabolic and Skeletal Defects of Infantile Hypophosphatasia. J. Bone Miner. Res. 1999, 14, 2015–2026.
- Whyte, M.P. Hypophosphatasia and the Role of Alkaline Phosphatase in Skeletal Mineralization. Endocr. Rev. 1994, 15, 439–461.
- Orimo, H. The Mechanism of Mineralization and the Role of Alkaline Phosphatase in Health and Disease. J. Nippon. Med. Sch. 2010, 77, 4–12.
- Harmey, D.; Hessle, L.; Narisawa, S.; Johnson, K.A.; Terkeltaub, R.; Millán, J.L. Concerted Regulation of Inorganic Pyrophosphate and Osteopontin by Akp2, Enpp1, and Ank. Am. J. Pathol. 2004, 164, 1199–1209.
- Whyte, M.P. Physiological role of alkaline phosphatase explored in hypophosphatasia. Ann. N. Y. Acad. Sci. 2010, 1192, 190–200.
- Waymire, K.G.; Mahuren, J.D.; Jaje, J.M.; Guilarte, T.R.; Coburn, S.P.; MacGregor, G.R. Mice lacking tissue non–specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat. Genet. 1995, 11, 45–51.
- Whyte, M.P.; Mahuren, J.D.; Vrabel, L.A.; Coburn, S.P. Markedly increased circulating pyridoxal-5’-phosphate levels in hypophosphatasia. Alkaline phosphatase acts in vitamin B6 metabolism. J. Clin. Investig. 1985, 76, 752–756.
- Brun-Heath, I.; Ermonval, M.; Chabrol, E.; Xiao, J.; Palkovits, M.; Lyck, R.; Miller, F.; Couraud, P.-O.; Mornet, E.; Fonta, C. Differential expression of the bone and the liver tissue non-specific alkaline phosphatase isoforms in brain tissues. Cell Tissue Res. 2010, 343, 521–536.
- Weiss, M.J.; Ray, K.; Henthorn, P.S.; Lamb, B.; Kadesch, T.; Harris, H. Structure of the human liver/bone/kidney alkaline phosphatase gene. J. Biol. Chem. 1988, 263, 12002–12010.
- Toh, Y.; Yamamoto, M.; Endo, H.; Misumi, Y.; Ikehara, Y. Isolation and characterization of a rat liver alkaline phosphatase gene. A single gene with two promoters. JBIC J. Biol. Inorg. Chem. 1989, 182, 231–237.
- Studer, M.; Terao, M.; Gianni, M.; Garattini, E. Characterization of a second promoter for the mouse liver/bone/kidney-type alkaline phosphatase gene: Cell and tissue specific expression. Biochem. Biophys. Res. Commun. 1991, 179, 1352–1360.
- Matsuura, S.; Kishi, F.; Kajii, T. Characterization of a 5′-flanking region of the human liver/bone/kidney alkaline phosphatase gene: Two kinds of mRNA from a single gene. Biochem. Biophys. Res. Commun. 1990, 168, 993–1000.
- Hahnel, A.; Rappolee, D.; Millan, J.; Manes, T.; Ziomek, C.; Theodosiou, N.; Werb, Z.; Pedersen, R.; Schultz, G. Two alkaline phosphatase genes are expressed during early development in the mouse embryo. Development 1990, 110, 555–564.
- Kruse, K.; Hanefeld, F.; Kohlschütter, A.; Rosskamp, R.; Gross-Selbeck, G. Hyperphosphatasia with mental retardation. J. Pediatr. 1988, 112, 436–439.
- Thompson, M.D.; Nezarati, M.M.; Gillessen-Kaesbach, G.; Meinecke, P.; Mendoza, R.; Mornet, E.; Brun-Heath, I.; Squarcioni, C.P.; Legeai-Mallet, L.; Munnich, A.; et al. Hyperphosphatasia with seizures, neurologic deficit, and characteristic facial features: Five new patients with Mabry syndrome. Am. J. Med. Genet. Part A 2010, 152A, 1661–1669.
- Thompson, M.D.; Cole, D.E.; Mabry, C.C. 50 Years Ago in T J P. J. Pediatr. 2020, 222, 97.
- Anstrom, J.A.; Brown, W.R.; Moody, D.M.; Thore, C.R.; Challa, V.R.; Block, S.M. Temporal expression pattern of cerebrovascular endothelial cell alkaline phosphatase during human gestation. J. Neuropathol. Exp. Neurol. 2002, 61, 76–84.
- Shimizu, N. Histochemical studies on the phosphatase of the nervous system. J. Comp. Neurol. 1950, 93, 201–217.
- Fonta, C.; Imbert, M. Vascularization in the Primate Visual Cortex during Development. Cereb. Cortex 2002, 12, 199–211.
- Bell, M.A.; Scarrow, W.G. Staining for microvascular alkaline phosphatase in thick celloidin sections of nervous tissue: Morphometric and pathological applications. Microvasc. Res. 1984, 27, 189–203.
- Norman, M.G.; O’Kusky, J.R. The growth and development of microvasculature in human cerebral cortex. J. Neuropathol. Exp. Neurol. 1986, 45, 222–232.
- Latker, C.H.; Shinowara, N.L.; Miller, J.C.; Rapoport, S.I. Differential localization of alkaline phosphatase in barrier tissues of the frog and rat nervous systems: A cytochemical and biochemical study. J. Comp. Neurol. 1987, 264, 291–302.
- Mizuguchi, H.; Hashioka, Y.; Utoguchi, N.; Kubo, K.; Nakagawa, S.; Mayumi, T. A Comparison of Drug Transport through Cultured Monolayers of Bovine Brain Capillary and Bovine Aortic Endothelial Cells. Biol. Pharm. Bull. 1994, 17, 1385–1390.
- Vorbrodt, A.W.; Lossinsky, A.S.; Wisniewski, H.M. Localization of Alkaline Phosphatase Activity in Endothelia of Developing and Mature Mouse Blood-Brain Barrier. Dev. Neurosci. 1986, 8, 1–13.
- Mayahara, H.; Hirano, H.; Saito, T.; Ogawa, K. The new lead citrate method for the ultracytochemical demonstration of activity of non-specific alkaline phosphatase (orthophosphoric monoester phosphohydrolase). Histochem. Cell Biol. 1967, 11, 88–96.
- Mori, S.; Nagano, M. Electron-microscopic cytochemistry of alkaline-phosphatase activity in endothelium, pericytes and oligodendrocytes in the rat brain. Histochem. Cell Biol. 1985, 82, 225–231.
- Ovtscharoff, W. Ultracytochemische Lokalisierung der alkalischen Phosphatase im Cortex cerebri bei neugeborenen Ratten. Histochem. Cell Biol. 1973, 37, 93–95.
- Deracinois, B.; Duban-Deweer, S.; Pottiez, G.; Cecchelli, R.; Karamanos, Y.; Flahaut, C. TNAP and EHD1 Are Over-Expressed in Bovine Brain Capillary Endothelial Cells after the Re-Induction of Blood-Brain Barrier Properties. PLoS ONE 2012, 7, e48428.
- Meyer, J.; Rauh, J.; Galla, H.-J. The Susceptibility of Cerebral Endothelial Cells to Astroglial Induction of Blood-Brain Barrier Enzymes Depends on Their Proliferative State. J. Neurochem. 1991, 57, 1971–1977.
- Rauh, J.; Meyer, J.; Beuckmann, C.; Galla, H.-J. Chapter 18: Development of an in vitro cell culture system to mimic the blood-brain barrier. Progress Brain Res. 1992, 91, 117–121.
- Tio, S.; Deenen, M.; Marani, E. Astrocyte-mediated induction of alkaline phosphatase activity in human umbilical cord vein endothelium: An in vitro model. Eur. J. Morphol. 1990, 28, 289–300.
- Ohlebusch, B.; Borst, A.; Frankenbach, T.; Klopocki, E.; Jakob, F.; Liedtke, D.; Graser, S. Investigation of alpl expression and Tnap-activity in zebrafish implies conserved functions during skeletal and neuronal development. Sci. Rep. 2020, 10, 1–16.
- McComb, R.B.; Bowers, G.N., Jr.; Posen, S. Alkaline Phosphatase; Springer Science and Business Media LLC: Berlin, Germany, 1979; 986p.
- Friede, R.L. A quantitative mapping of alkaline phosphatase in the brain of the rhesus monkey. J. Neurochem. 1966, 13, 197–203.
- Negyessy, L.; Xiao, J.; Kántor, O.; Kovacs, G.G.; Palkovits, M.; Doczi, T.; Renaud, L.; Baksa, G.; Glasz, T.; Ashaber, M.; et al. Layer-specific activity of tissue non-specific alkaline phosphatase in the human neocortex. Neuroscience 2011, 172, 406–418.
- Liu, J.; Nam, H.K.; Campbell, C.; Gasque, K.C.D.S.; Millán, J.L.; Hatch, N.E. Tissue-nonspecific alkaline phosphatase deficiency causes abnormal craniofacial bone development in the Alpl−/− mouse model of infantile hypophosphatasia. Bone 2014, 67, 81–94.
- Liedtke, D.; Hofmann, C.; Jakob, F.; Klopocki, E.; Graser, S. Tissue-Nonspecific Alkaline Phosphatase—A Gatekeeper of Physiological Conditions in Health and a Modulator of Biological Environments in Disease. Biomolecules 2020, 10, 1648.
- Sebastián-Serrano, Á.; De Diego-García, L.; Henshall, D.C.; Engel, T.; Diaz-Hernandez, M. Haploinsufficient TNAP Mice Display Decreased Extracellular ATP Levels and Expression of Pannexin-1 Channels. Front. Pharmacol. 2018, 9.
- Gámez-Belmonte, R.; Tena-Garitaonaindia, M.; Hernández-Chirlaque, C.; Córdova, S.; Ceacero-Heras, D.; De Medina, F.S.; Martínez-Augustin, O. Deficiency in Tissue Non-Specific Alkaline Phosphatase Leads to Steatohepatitis in Mice Fed a High Fat Diet Similar to That Produced by a Methionine and Choline Deficient Diet. Int. J. Mol. Sci. 2020, 22, 51.
- Hernández-Chirlaque, C.; Gámez-Belmonte, R.; Ocón, B.; Martínez-Moya, P.; Wirtz, S.; de Medina, F.S.; Martínez-Augustin, O. Tissue Non-specific Alkaline Phosphatase Expression is Needed for the Full Stimulation of T Cells and T Cell-Dependent Colitis. J. Crohns Colitis 2016, 11, 857–870.
- Yamamoto, S.; Orimo, H.; Matsumoto, T.; Iijima, O.; Narisawa, S.; Maeda, T.; Millán, J.L.; Shimada, T. Prolonged survival and phenotypic correction of Akp2−/− hypophosphatasia mice by lentiviral gene therapy. J. Bone Miner. Res. 2010, 26, 135–142.
- Nam, H.K.; Vesela, I.; Schutte, S.D.; Hatch, N.E. Viral delivery of tissue nonspecific alkaline phosphatase diminishes craniosynostosis in one of two FGFR2C342Y/+ mouse models of Crouzon syndrome. PLoS ONE 2020, 15, e0234073.
- Arnò, B.; Galli, F.; Roostalu, U.; Aldeiri, B.M.; Miyake, T.; Albertini, A.; Bragg, L.; Prehar, S.; McDermott, J.C.; Cartwright, E.J.; et al. TNAP limits TGF-β-dependent cardiac and skeletal muscle fibrosis by inactivating the SMAD2/3 transcription factors. J. Cell Sci. 2019, 132, jcs.234948.
- Sheen, C.R.; Kuss, P.; Narisawa, S.; Yadav, M.C.; Nigro, J.; Wang, W.; Chhea, T.N.; Sergienko, E.A.; Kapoor, K.; Jackson, M.R.; et al. Pathophysiological Role of Vascular Smooth Muscle Alkaline Phosphatase in Medial Artery Calcification. J. Bone Miner. Res. 2015, 30, 824–836.
- Savinov, A.Y.; Salehi, M.; Yadav, M.C.; Radichev, I.A.; Millán, J.L.; Savinova, O.V. Transgenic Overexpression of Tissue-Nonspecific Alkaline Phosphatase (TNAP) in Vascular Endothelium Results in Generalized Arterial Calcification. J. Am. Hear. Assoc. 2015, 4, e002499.
- Rodionov, R.N.; Begmatov, H.; Jarzebska, N.; Patel, K.; Mills, M.T.; Ghani, Z.; Khakshour, D.; Tamboli, P.; Patel, M.N.; Abdalla, M.; et al. Homoarginine Supplementation Prevents Left Ventricular Dilatation and Preserves Systolic Function in a Model of Coronary Artery Disease. J. Am. Hear. Assoc. 2019, 8, e012486.
- Romanelli, F.; Corbo, A.; Salehi, M.; Yadav, M.C.; Salman, S.; Petrosian, D.; Rashidbaigi, O.J.; Chait, J.; Kuruvilla, J.; Plummer, M.; et al. Overexpression of tissue-nonspecific alkaline phosphatase (TNAP) in endothelial cells accelerates coronary artery disease in a mouse model of familial hypercholesterolemia. PLoS ONE 2017, 12, e0186426.
- Brichacek, A.L.; Benkovic, S.A.; Chakraborty, S.; Nwafor, D.C.; Wang, W.; Jun, S.; Dakhlallah, D.; Geldenhuys, W.J.; Pinkerton, A.B.; Millán, J.L.; et al. Systemic inhibition of tissue-nonspecific alkaline phosphatase alters the brain-immune axis in experimental sepsis. Sci. Rep. 2019, 9, 1–19.
- Foster, B.; Kuss, P.; Yadav, M.; Kolli, T.; Narisawa, S.; Lukashova, L.; Cory, E.; Sah, R.; Somerman, M.; Millán, J. Conditional Alpl Ablation Phenocopies Dental Defects of Hypophosphatasia. J. Dent. Res. 2016, 96, 81–91.
- Nwafor, D.C.; Brichacek, A.L.; Wang, W.; Bidwai, N.; Lilly, C.L.; Millan, J.; Brown, C.M. Brain endothelial cell tissue-nonspecific alkaline phosphatase (TNAP) activity promotes maintenance of barrier integrity via the ROCK pathway. bioRxiv 2021.
- Alva, J.A.; Zovein, A.C.; Monvoisin, A.; Murphy, T.; Salazar, A.; Harvey, N.L.; Carmeliet, P.; Iruela-Arispe, M.L. VE-Cadherin-Cre-recombinase transgenic mouse: A tool for lineage analysis and gene deletion in endothelial cells. Dev. Dyn. 2006, 235, 759–767.
- Assmann, J.C.; Körbelin, J.; Schwaninger, M. Genetic manipulation of brain endothelial cells in vivo. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2016, 1862, 381–394.
- Rufo, M.B.; Fishman, W.H. l-Homoarginine, a specific inhibitor of liver-type alkaline phosphatase, applied to the recognition of liver-type enzyme activity in rat intestine. J. Histochem. Cytochem. 1972, 20, 336–343.
- Kozlenkov, A.; Le Du, M.H.; Cuniasse, P.; Ny, T.; Hoylaerts, M.F.; Millán, J.L. Residues Determining the Binding Specificity of Uncompetitive Inhibitors to Tissue-Nonspecific Alkaline Phosphatase. J. Bone Miner. Res. 2004, 19, 1862–1872.
- Deracinois, B.; Lenfant, A.-M.; Dehouck, M.-P.; Flahaut, C. Tissue Non-specific Alkaline Phosphatase (TNAP) in Vessels of the Brain. Subcell. Biochem. 2015, 76, 125–151.
- Dahl, R.; Sergienko, E.A.; Su, Y.; Mostofi, Y.S.; Yang, L.; Simão, A.M.; Narisawa, S.; Brown, B.; Mangravita-Novo, A.; Vicchiarelli, M.; et al. Discovery and Validation of a Series of Aryl Sulfonamides as Selective Inhibitors of Tissue-Nonspecific Alkaline Phosphatase (TNAP). J. Med. Chem. 2009, 52, 6919–6925.
- Nakamura, T.; Nakamura-Takahashi, A.; Kasahara, M.; Yamaguchi, A.; Azuma, T. Tissue-nonspecific alkaline phosphatase promotes the osteogenic differentiation of osteoprogenitor cells. Biochem. Biophys. Res. Commun. 2020, 524, 702–709.
- Pinkerton, A.B.; Sergienko, E.; Bravo, Y.; Dahl, R.; Ma, C.-T.; Sun, Q.; Jackson, M.R.; Cosford, N.D.P.; Millán, J.L. Discovery of 5-((5-chloro-2-methoxyphenyl)sulfonamido)nicotinamide (SBI-425), a potent and orally bioavailable tissue-nonspecific alkaline phosphatase (TNAP) inhibitor. Bioorg. Med. Chem. Lett. 2018, 28, 31–34.
- Genest, F.; Rak, D.; Petryk, A.; Seefried, L. Physical Function and Health-Related Quality of Life in Adults Treated With Asfotase Alfa for Pediatric-Onset Hypophosphatasia. JBMR Plus 2020, 4.
- Kishnani, P.S.; Rockman-Greenberg, C.; Rauch, F.; Bhatti, M.T.; Moseley, S.; Denker, A.E.; Watsky, E.; Whyte, M.P. Five-year efficacy and safety of asfotase alfa therapy for adults and adolescents with hypophosphatasia. Bone 2019, 121, 149–162.
- Whyte, M.P.; Simmons, J.H.; Moseley, S.; Fujita, K.P.; Bishop, N.; Salman, N.J.; Taylor, J.; Phillips, D.; McGinn, M.; McAlister, W.H. Asfotase alfa for infants and young children with hypophosphatasia: 7 year outcomes of a single-arm, open-label, phase 2 extension trial. Lancet Diabetes Endocrinol. 2019, 7, 93–105.
- Pickkers, P.; Snellen, F.; Rogiers, P.; Bakker, J.; Jorens, P.; Meulenbelt, J.; Spapen, H.; Tulleken, J.E.; Lins, R.; Ramael, S.; et al. Clinical pharmacology of exogenously administered alkaline phosphatase. Eur. J. Clin. Pharmacol. 2009, 65, 393–402.
- Heemskerk, S.; Masereeuw, R.; Moesker, O.; Bouw, M.P.W.J.M.; Van Der Hoeven, J.G.; Peters, W.H.M.; Russel, F.G.M.; Pickkers, P. Alkaline phosphatase treatment improves renal function in severe sepsis or septic shock patients. Crit. Care Med. 2009, 37, 417-e1.
- Bender, B.; Baranyi, M.; Kerekes, A.; Bodrogi, L.; Brands, R.; Uhrin, P.; Bösze, Z. Recombinant Human Tissue Non-Specific Alkaline Phosphatase Successfully Counteracts Lipopolysaccharide Induced Sepsis in Mice. Physiol. Res. 2015, 64, 731–738.
- Pickkers, P.; Heemskerk, S.; Schouten, J.; Laterre, P.-F.; Vincent, J.-L.; Beishuizen, A.; Jorens, P.G.; Spapen, H.; Bulitta, M.; Peters, W.H.; et al. Alkaline phosphatase for treatment of sepsis-induced acute kidney injury: A prospective randomized double-blind placebo-controlled trial. Crit. Care 2012, 16, R14.
- Lukas, M.; Drastich, P.; Konecny, M.; Gionchetti, P.; Urban, O.; Cantoni, F.; Bortlik, M.; Duricova, D.; Bulitta, M. Exogenous alkaline phosphatase for the treatment of patients with moderate to severe ulcerative colitis. Inflamm. Bowel Dis. 2010, 16, 1180–1186.
- Engelmann, C.; Adebayo, D.; Oria, M.; De Chiara, F.; Novelli, S.; Habtesion, A.; Davies, N.; Andreola, F.; Jalan, R. Recombinant Alkaline Phosphatase Prevents Acute on Chronic Liver Failure. Sci. Rep. 2020, 10, 1–13.
- Peters, E.; Ergin, B.; Kandil, A.; Gurel-Gurevin, E.; Van Elsas, A.; Masereeuw, R.; Pickkers, P.; Ince, C. Effects of a human recombinant alkaline phosphatase on renal hemodynamics, oxygenation and inflammation in two models of acute kidney injury. Toxicol. Appl. Pharmacol. 2016, 313, 88–96.
- Juschten, J.; for the BASIC study investigators; Ingelse, S.A.; Bos, L.D.J.; Girbes, A.R.J.; Juffermans, N.P.; Van Der Poll, T.; Schultz, M.J.; Tuinman, P.R. Alkaline phosphatase in pulmonary inflammation—A translational study in ventilated critically ill patients and rats. Intensive Care Med. Exp. 2020, 8, 1–14.
- Pickkers, P.; Mehta, R.L.; Murray, P.T.; Joannidis, M.; Molitoris, B.A.; Kellum, J.A.; Bachler, M.; Hoste, E.A.J.; Hoiting, O.; Krell, K.; et al. Effect of Human Recombinant Alkaline Phosphatase on 7-Day Creatinine Clearance in Patients With Sepsis-Associated Acute Kidney Injury. JAMA 2018, 320, 1998–2009.
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L.; et al. A molecular atlas of cell types and zonation in the brain vasculature. Nat. Cell Biol. 2018, 554, 475–480.
- Yang, A.C.; Stevens, M.Y.; Chen, M.B.; Lee, D.P.; Stähli, D.; Gate, D.; Contrepois, K.; Chen, W.; Iram, T.; Zhang, L.; et al. Physiological blood–brain transport is impaired with age by a shift in transcytosis. Nat. Cell Biol. 2020, 583, 425–430.
- Beuckmann, C.; Hellwig, S.; Galla, H.-J. Induction of the Blood/Brain-Barrier-Associated Enzyme Alkaline Phosphatase in Endothelial Cells from Cerebral Capillaries is Mediated Via cAMP. JBIC J. Biol. Inorg. Chem. 1995, 229, 641–644.
- Roux, F.; Durieu-Trautmann, O.; Chaverot, N.; Claire, M.; Mailly, P.; Bourre, J.-M.; Strosberg, A.D.; Couraud, P.-O. Regulation of gamma-glutamyl transpeptidase and alkaline phosphatase activities in immortalized rat brain microvessel endothelial cells. J. Cell. Physiol. 1994, 159, 101–113.
- Shibuya, M.; Hirai, S.; Seto, M.; Satoh, S.-I.; Ohtomo, E. Effects of fasudil in acute ischemic stroke: Results of a prospective placebo-controlled double-blind trial. J. Neurol. Sci. 2005, 238, 31–39.
- Fukuta, T.; Asai, T.; Yanagida, Y.; Namba, M.; Koide, H.; Shimizu, K.; Oku, N. Combination therapy with liposomal neuroprotectants and tissue plasminogen activator for treatment of ischemic stroke. FASEB J. 2017, 31, 1879–1890.
- Liu, K.; Li, Z.; Wu, T.; Ding, S. Role of Rho Kinase in Microvascular Damage Following Cerebral Ischemia Reperfusion in Rats. Int. J. Mol. Sci. 2011, 12, 1222–1231.
- Jianjun, Z.; Baochun, Z.; Limei, M.; Lijun, L. Exploring the beneficial role of ROCK inhibitors in sepsis-induced cerebral and cognitive injury in rats. Fundam. Clin. Pharmacol. 2021.
- Adams, S.E.; Melnykovych, G. Synergistic stimulation of alkaline phosphatase activity in bovine aortic endothelial cells grown in the presence of retinoids and glucocorticoids. J. Cell. Physiol. 1985, 124, 120–124.
- Nakazato, H.; Deguchi, M.; Fujimoto, M.; Fukushima, H. Alkaline phosphatase expression in cultured endothelial cells of aorta and brain micro vessels: Induction by interleukin-6-type cytokines and suppression by transforming growth factor betas. Life Sci. 1997, 61, 2065–2072.
- Romero, C.R.; Herzig, D.S.; Etogo, A.; Nunez, J.; Mahmoudizad, R.; Fang, G.; Murphey, E.D.; Toliver-Kinsky, T.; Sherwood, E.R. The role of interferon-γ in the pathogenesis of acute intra-abdominal sepsis. J. Leukoc. Biol. 2010, 88, 725–735.
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12.
- Brown, W.R.; Moody, D.M.; Thore, C.R.; Challa, V.R.; Anstrom, J.A. Vascular dementia in leukoaraiosis may be a consequence of capillary loss not only in the lesions, but in normal-appearing white matter and cortex as well. J. Neurol. Sci. 2007, 257, 62–66.
- Goncharov, N.V.; Nadeev, A.D.; Jenkins, R.O.; Avdonin, P.V. Markers and Biomarkers of Endothelium: When Something Is Rotten in the State. Oxid. Med. Cell. Longev. 2017, 2017, 1–27.
- Nwafor, D.C.; Brichacek, A.L.; Mohammad, A.S.; Griffith, J.; Lucke-Wold, B.P.; Benkovic, S.A.; Geldenhuys, W.J.; Lockman, P.R.; Brown, C.M. Targeting the Blood-Brain Barrier to Prevent Sepsis-Associated Cognitive Impairment. J. Central Nerv. Syst. Dis. 2019, 11.
- Nwafor, D.C.; Chakraborty, S.; Brichacek, A.L.; Jun, S.; Gambill, C.A.; Wang, W.; Engler-Chiurazzi, E.B.; Dakhlallah, D.; Pinkerton, A.B.; Millán, J.L.; et al. Loss of tissue-nonspecific alkaline phosphatase (TNAP) enzyme activity in cerebral microvessels is coupled to persistent neuroinflammation and behavioral deficits in late sepsis. Brain Behav. Immun. 2020, 84, 115–131.
- Iwashyna, T.J.; Ely, E.W.; Smith, D.M.; Langa, K.M. Long-term Cognitive Impairment and Functional Disability Among Survivors of Severe Sepsis. JAMA 2010, 304, 1787–1794.
- Andonegui, G.; Zelinski, E.L.; Schubert, C.L.; Knight, D.; Craig, L.A.; Winston, B.W.; Spanswick, S.C.; Petri, B.; Jenne, C.N.; Sutherland, J.C.; et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight 2018, 3.
- Chavan, S.S.; Huerta, P.T.; Robbiati, S.; Valdes-Ferrer, S.I.; Ochani, M.; Dancho, M.; Frankfurt, M.; Volpe, B.T.; Tracey, K.J.; Diamond, B. HMGB1 Mediates Cognitive Impairment in Sepsis Survivors. Mol. Med. 2012, 18, 930–937.
- Barone, F.C.; Arvin, B.; White, R.F.; Miller, A.R.; Webb, C.L.; Willette, R.N.; Lysko, P.G.; Feuerstein, G.Z. Tumor Necrosis Factor-α. Stroke 1997, 28, 1233–1244.
- Michie, H.R.; Manogue, K.R.; Spriggs, D.R.; Revhaug, A.; O’Dwyer, S.; Dinarello, C.A.; Cerami, A.; Wolff, S.M.; Wilmore, D.W. Detection of Circulating Tumor Necrosis Factor after Endotoxin Administration. N. Engl. J. Med. 1988, 318, 1481–1486.
- Yilmaz, G.; Arumugam, T.V.; Stokes, K.Y.; Granger, D.N. Role of T Lymphocytes and Interferon-γ in Ischemic Stroke. Circulation 2006, 113, 2105–2112.
- Colton, C.A.; Wilcock, D.M.; Wink, D.A.; Davis, J.; Van Nostrand, W.E.; Vitek, M.P. The Effects of NOS2 Gene Deletion on Mice Expressing Mutated Human AβPP. J. Alzheimer’s Dis. 2008, 15, 571–587.
- Wilcock, D.M.; Lewis, M.R.; Van Nostrand, W.E.; Davis, J.; Previti, M.L.; Gharkholonarehe, N.; Vitek, M.P.; Colton, C.A. Progression of Amyloid Pathology to Alzheimer’s Disease Pathology in an Amyloid Precursor Protein Transgenic Mouse Model by Removal of Nitric Oxide Synthase 2. J. Neurosci. 2008, 28, 1537–1545.
- Nwafor, D.C.; Chakraborty, S.; Jun, S.; Brichacek, A.L.; Dransfeld, M.; Gemoets, D.E.; Dakhlallah, D.; Brown, C.M. Disruption of metabolic, sleep, and sensorimotor functional outcomes in a female transgenic mouse model of Alzheimer’s disease. Behav. Brain Res. 2021, 398, 112983.
- Díaz-Hernández, M.; Gómez-Ramos, A.; Rubio, A.; Gómez-Villafuertes, R.; Naranjo, J.R.; Miras-Portugal, M.T.; Avila, J. Tissue-nonspecific Alkaline Phosphatase Promotes the Neurotoxicity Effect of Extracellular Tau. J. Biol. Chem. 2010, 285, 32539–32548.
- Banks, W.A.; Reed, M.J.; Logsdon, A.F.; Rhea, E.M.; Erickson, M.A. Healthy aging and the blood–brain barrier. Nat. Aging 2021, 1, 243–254.
- Yousef, H.; Czupalla, C.J.; Lee, D.; Chen, M.B.; Burke, A.N.; Zera, K.A.; Zandstra, J.; Berber, E.; Lehallier, B.; Mathur, V.; et al. Aged blood impairs hippocampal neural precursor activity and activates microglia via brain endothelial cell VCAM1. Nat. Med. 2019, 25, 988–1000.
- Chen, M.B.; Yang, A.C.; Yousef, H.; Lee, D.; Chen, W.; Schaum, N.; Lehallier, B.; Quake, S.R.; Wyss-Coray, T. Brain Endothelial Cells Are Exquisite Sensors of Age-Related Circulatory Cues. Cell Rep. 2020, 30, 4418–4432.


