 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Álvaro Del Río García | + 1638 word(s) | 1638 | 2021-06-03 08:24:42 | | | |
| 2 | Nora Tang | Meta information modification | 1638 | 2021-06-07 03:06:13 | | | | |
| 3 | Isabel Rodríguez | + 1096 word(s) | 2734 | 2021-06-07 17:00:29 | | |
Video Upload Options
Bicuspid aortic valve (BAV) associated with aortopathy is the most common congenital heart disease in the general population. Far from being a simple harmless valve malformation, it can be a complex and heterogeneous disease and a source of chronic and acute pathology (early valvular disease, aneurysm, dissection). In the previous years, intense research has been carried out to find out and understand its mechanisms, but the pathophysiology of the disease is still not fully understood and many questions remain open. Recent studies have discovered several genetic mutations involved in the development of valvular and aortic malformations, but still cannot explain more than 5–10% of cases. Other studies have also focused on molecular alterations and cellular processes (TGF-β pathway, microRNAs, degradation of the extracellular matrix, metalloproteinases, etc.), being a field in constant search and development, looking for a therapeutic target to prevent the development of the disease. Increased knowledge about this multifaceted disorder, derived from both basic and clinical research, may influence the diagnosis, follow-up, prognosis, and therapies of affected patients in the near future.
1. Introduction
It has long been known that the bicuspid aortic valve (BAV) is the most common congenital heart defect, with a prevalence differing according to studies (0.5–2% of births) [1][2]. With many cases in young people, it generates a high annual morbidity and mortality, derived not only from valve dysfunction (stenosis due to early calcification or aortic regurgitation) but also from aortic complications (Figure 1). It has been shown, depending on the reports, that up to 35–80% of patients with BAV associate dilation of the ascending aorta [3][4]. Therefore, the patients have a higher age-adjusted relative risk of aortic dissection than the general population. Given the close relationship between valve alteration and its aortic continuity, the term “bicuspid aortopathy” has begun to be used in the literature [5].
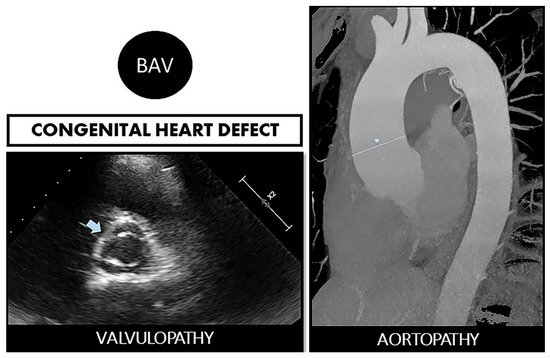
Figure 1. Bicuspid aortopathy is a double road. The congenital defect can develop in the person who suffers from both valvulopathy (stenosis or regurgitation) and aortopathy (aneurysms and risk of spontaneous dissection). On the left, echocardiographic image of BAV (arrow). On the right, a resonance image of dilated ascending aorta (marked straight line).
Classically, an autosomal dominant inheritance has been described, but appears with reduced penetrance (same genetic variant but no disease) and variable expressivity (same genetic variant with different manifestations of disease) [6]. In any case, it is recognized that screening in family of the index case is necessary because the prevalence of BAV in first-degree relatives is 10 times higher than that in the general population [7].
Since only symptomatic patients seek medical attention, there is a knowledge gap about the real consequences of having BAV, and thus, many patients remain underdiagnosed. BAV is generally diagnosed in adulthood with the onset of aortic valvular dysfunction [8]. Practicable methods include both transthoracic (TTE) and transesophageal echocardiography (TEE), cardiac computed tomography (CT), and cardiac magnetic resonance (CMR).
More than 50% of patients with BAV undergo aortic valve replacement during their lifetime, and more than 25% of patients with BAV undergo aortic surgery performed for dilation of the aortic root or ascending aorta, often concurrent with aortic valve replacement [4][9].
In the last decade, many studies have been published focusing on the finding genetic and molecular alterations present in families with BAV and consistent inheritance, through registries of patients with BAV and tricuspid aortic valve (TAV), looking for differences between them [10]. Isolated mutations, several genes, cellular pathways, different microRNAs, histopathological alterations, etc. have been found associated with the bicuspid aortopathy. Many pieces of this immense puzzle are unraveled in the following pages, being aware that in the coming years, the task will consist of interconnecting these elements. Consequently, we can say that despite intense research in the recent years, the manifest genetic, epigenetic, and molecular heterogeneity and complexity of this pathology means that the final pathogenic and ontogenetic mechanisms are still not fully understood. This article reviews the latest and outstanding advances in molecular and genetic investigations on bicuspid aortopathy.
2. BAV and Aortopathy: A Two-Way Road
The etiology and pathogenesis that lead to dilation of the ascending aorta in patients with a BAV are uncertain to date. The actual incidence and prevalence of aortopathy are not truly known either, since it is often an alteration with an asymptomatic course. Although, as classic studies explain, aortic dilation is a relatively common complication in patients with BAV, appearing at an earlier age and progressing continuously and rather faster [11][12]. Two theories have been proposed, two non-exclusive roads.
One of the roads is the “genetic theory”. As we have explained later, this theory defends the existence of mutations in different genes in the patients who carry a double leaflet valve and that affect the aorta at the same time. This connection should not be surprising since the aortic valve and the ascending aorta share the same embryological origin [13].
The BAV disease is considered nowadays as an autosomal dominant disorder with low penetrance and variable expressivity. Although different genes with divergent inheritance pattern have been associated to this entity, its genetic bases are still not completely known [14]. BAV can present as an isolated condition or associated with syndromic disorders such as Marfan [15] and Turner syndromes [16] and with other cardiovascular diseases such as coarctation of aorta and ventricular septal defect. Familial clustering was initially suggested in different studies [17][18] and the first estimation of its heritability was proposed by Cripe et al. [19]. Here, we review the genetic basis of BAV aortopathy and some of the candidate genes that have been implicated in the process (Table 1).
Table 1. Main genes associated with bicuspid aortopathy.
|
Gene |
OMIM |
Pathway/Role |
Mutation Consequence |
Reference |
|
NOTCH1 |
190198 |
Development of valve and endocardial cushions |
Defect on the aortic valve and de-repression of calcium deposition |
|
|
GATA4 |
600576 |
Cardiovascular embryogenesis |
Enhanced susceptibility to BAV |
|
|
GATA5 |
611496 |
Cardiovascular embryogenesis |
BAV development |
|
|
GATA6 |
601656 |
Cardiovascular embryogenesis |
Enhanced susceptibility to familial BAV |
|
|
TBX5 |
601620 |
Cardiac development |
Atrial fibrillation. Segregates with BAV |
[27] |
|
SMAD6 |
602931 |
TGF-β signaling pathway |
Related with BAV + aortic aneurysm |
|
|
FBN1 |
134797 |
Integrity of the aortic media |
Related with isolated BAV and Marfan syndrome with aortic root dilation |
[30] |
|
ROBO4 |
607528 |
Transmembrane receptor |
Segregating with ascending aortic aneurysm in BAV |
[31] |
|
ACTA2 |
102620 |
Smooth muscle α-actin |
Related with familial ascending aortic aneurysm in BAV |
[32] |
|
ELN |
130160 |
Arterial development |
Correlated with ascending aorta diameter in BAV |
[33] |
The main gene with the strongest evidence that has been demonstrated to be associated with BAV in both familial and sporadic forms is NOTCH1 (9q 34.4, OMIM 190198). The NOTCH1 pathway is directly involved in the development of the valve-forming fields during cardiogenesis and is crucial for the correct development of endocardial cushions through regulation of endocardial epithelial-to-mesenchymal transition and remodeling of the immature aortic valve [20]. Mutations in NOTCH1 cause an early developmental defect in the aortic valve and a later de-repression of calcium deposition that causes progressive aortic valve disease [21]. Besides, loss-of-function mutations in NOTCH1 have also been associated with other congenital heart diseases involving the left ventricle outflow tract, mitral valve, and, less frequently, with right heart cardiac lesions, in an autosomal dominant trait with variable expressivity [34]. Otherwise, thoracic aortic aneurysms have also been described in pedigrees with inactivating NOTCH1 mutations. Thus, the study published by Kerstjens-Frederikse et al., determining the prevalence and spectrum of NOTCH1 mutations in left-sided congenital heart disease, found that thoracic aortic aneurysms occurred in six mutation carriers [35].
Furthermore, members of the GATA family of the zinc finger superfamily of transcriptions factors have also been related to BAV disease [36]. This family consists of six members divided in two subgroups based on their sequence homology and tissue location: GATA1, 2, and 3 are mainly implicated in hematopoietic development and GATA4, 5, and 6 are essential in cardiovascular embryogenesis, as well as in other tissues derived from mesoderm and endoderm. Accordingly, a GATA4 (8p23.1, OMIM 600576) loss-of-function mutation has been associated with enhanced susceptibility to BAV [22]. GATA5 (20q13.33, OMIM 611496) has been demonstrated to be involved in cardiac morphogenesis and aortic valve development implicated in BAV pathogenesis [24]. Our group have previously identified four genetic variants in GATA4, GATA5, and GATA6 (18q11.2, OMIM 601656) only present in patients with BAV, with a potential pathogenic effect predicted by bioinformatic tools, supporting the implication of these genes in the development of this valvulopathy [23]. A GATA6 loss-of-function mutation has also been associated with enhanced susceptibility to familial BAV and also recent studies suggest that haploinsufficiency of transcription factor GATA6 leads to BAV [25][26].
Another cardiac transcription factor recently associated with BAV is TBX5 (12q24.21, OMIM 601620). This gene, previously associated with atrial fibrillation, was sequenced in unrelated adult patients suffering from both congenital heart defects and atrial fibrillation. A novel nonsense mutation that impairs the transcriptional activity of the protein was found segregating also with BAV in the family [27]. This finding highlights the importance of deciphering the molecular pathways implicated in order to identify new candidate genes for genetic studies.
The transcriptional regulator encoded by SMAD6 gene (15q22.31, OMIM 602931) has also been confirmed to be related to BAV and associated thoracic aortic aneurysm [28]. Luyckx et al. identified seven novel likely pathogenic SMAD6 variants in BAV individuals with thoracic aortic aneurysm, establishing the role of SMAD6 variants in their etiology and revealing limited contribution to thoracic aneurysms development in patients with TAV. Familial segregation studies confirmed reduced penetrance and variable clinical expressivity. The results of these studies improved insights into the clinical spectrum of SMAD6-related BAV-thoracic aneurysms [29].
FBN1 gene (15q21.1, OMIM 134797) encodes an extracellular glycoprotein (fibrillin-1), component of connective tissue that is secreted by vascular smooth muscle cells, and regulates the structural integrity of the aortic media. Previous studies have evidenced the presence of genetic variants in FBN1 gene in isolated BAV patients with aortic root dilation and in Marfan syndrome patients with BAV [30]. Regarding the superior prevalence of BAV in patients with Marfan syndrome to the general population [15], common underlying mechanisms for these two entities have been proposed: increased metalloproteinase (MMP) activity and a decreased fibrillin-1 expression in the aortic wall [37]. Moreover, fibrillin-1 deficiency could be responsible for matrix alterations, which contribute to aortopathy associated with BAV without Marfan syndrome [38].
Another candidate gene is ROBO4 (11q24.2, OMIM 607528) in which Gould et al. found two mutations segregating with ascending aortic aneurysm in BAV subjects in two families. The involvement of ROBO4 in BAV formation and in BAV-associated ascending aortic aneurysm was confirmed using animals deficient for Robo4 as well as by endothelial cell-specific Robo4 silencing or mutation [31].
Finally, missense mutations in ACTA2 gene (10q23.31, OMIM 102620), encoding smooth muscle α-actin, had been shown to be responsible for 14% of familial ascending aortic aneurysms and dissections [32]. However, this gene, although candidate, does not seem to play a significant role in the pathogenesis of BAV aortopathy as concluded by Tortora et al. in a small patient cohort [39].
Not only mutations but also polymorphisms could prove important in the early identification of the patients at higher risk. The functional polymorphism rs2071307 in the elastin gene (ELN, 7q11.23, OMIM 130160) has been shown to correlate with circulating elastin soluble fragments and ascending aorta diameter in BAV patients [33]. These facts could be an important starting point in the explanation of aneurysm formation in these subjects, and future studies will have to show whether increased spatial heterogeneity and temporal dispersion of aortic strain distribution might be specific signs of aneurysm formation.
As a conclusion, and as Messner et al. reflect in their recent review, although there is a large list of genes linked to BAV and BAV-aortopathy, we are still far from a complete knowledge of this complex and heterogeneous disease [40]. Many “genetic” questions still remain.
The other road is the “hemodynamic theory”. Due to the anomalous opening of the BAV, an eccentric flow is formed directed against the aortic wall, this constant tangential force being the one that ends up generating the dilation. The consequent load is known as “wall shear stress” (WSS) [41].
In the past decade, using 4D flow cardiovascular magnetic resonance (CMR 4D), it was found that the flow patterns in the ascending aorta of the patients with RL-BAV (fusion subtype of the right and left coronary cusps) and RN-BAV (fusion subtype of the right and non-coronary cusps) were different: RL-BAV impacted the anterior aortic wall and RN-BAV influenced directly on its posterior side, explaining different aortic dilation morphotypes [42][43]. However, there are also studies reporting a weak or no association between the BAV morphotype and the location of the aneurysm [44][45]. To elucidate these knowledge gaps, many subsequent studies continue to emerge in the recent years in this field. However, the subtypes according to the location of the raphe affect aortic flow. Moreover, valve degeneration has been shown to affect flow and WSS, being more severe and rotational in the patients with severe stenosis independently of valve morphology [46].
It has been suggested in BAV that aortic dilation development is an adaptive mechanism of the arterial wall. A remodeling through mechano-sensing is possible. Regions with increased WSS have an extracellular matrix dysregulation and elastic fiber degeneration, with higher levels of MMP-2, MMP-3, and TGF-β in the areas with the higher WSS, associated with a greater loss of architecture in the middle layer [47][48]. It has also been observed that patients with BAV-aortic stenosis present increased WSS and thinning and loss of elastic fiber architecture more markedly than those with BAV-aortic regurgitation. In addition, the regional histopathology alteration manifests itself more strongly in mildly aortic dilation (<4.5 cm) [49][50].
However, not all patients with the same valvular phenotype have the same pattern of aortopathy and, furthermore, 25–35% of BAV present a non-dilated aorta in the follow-up [3][51]. However, controversies continue to exist. For example, first-degree relatives (with TAV) of patients with BAV suffer more frequently from aortic aneurysms, without presenting these eccentric flows involving WSS [52]. In a recent meta-analysis by Girdauskas et al., the appearance of aortic dissection on post-aortic valve replacement was evaluated in the patients with BAV. It was found that those with regurgitation had a higher risk of dissection at follow-up, which could indicate greater tissue weakness. This occurs, even with the new valve replaced, where the WSS secondary to the altered outflow is not present. In contrast, BAV stenosis-associated aortopathy seems to follow a more benign course post-aortic valve replacement [53].
Other non-hemodynamics factors must be linked. Currently, the results do not clearly indicate that hemodynamic effects on the aorta, due to outflow eccentricity, explain bicuspid aneurysms by themselves. Longer follow-up records (even decades) of patients with BAV are necessary to achieve clarity about the relationship of WSS and aneurysms, especially in non-dysfunctional BAVs.
It seems more sensible to think that the two phenomena (genetics + hemodynamics) participate. The impaired aortic ejection flow, with a weak arterial wall that is already genetically determined or histo-molecularly altered, will possibly cause or accelerate the development of aneurysms (Figure 2). Understanding the mechanisms underlying vascular remodeling of the aortic wall is essential for the development of new risk scales, not based on measurements of aortic size, but on imaging parameters or biomarkers, which allow establishing the best individualized surgical time.
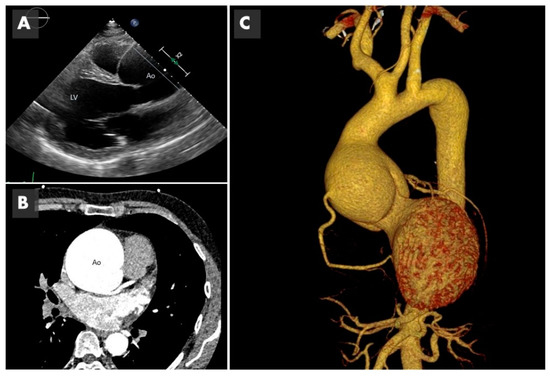
Figure 2. Imaging of bicuspid associated aneurysm. (A) The 2D image of a transthoracic echocardiogram in the parasternal long axis plane showing the dilated aorta (Ao) marked with the straight line (LV: left ventricle). (B) Computed tomography (CT) in axial plane showing the very dilated ascending aorta (Ao) at the level of the sinuses of Valsalva. (C) The 3D CT reconstruction, showing a huge 8 cm aneurysm.
3. Future Directions
The search for the ultimate genetic or epigenetic cause of the different bicuspid phenotypes should be facilitated by the next-generation sequencing tools that allow study of large populations at low cost.
In the near future, studies should also be directed to improve diagnostic and stratification criteria to better predict a more precise personalized risk for BAV patients. To achieve this proposal, imaging methods around the use of modern techniques, such as CMR-4D, are essential [54].
The novel field of the ncRNAs requires further studies to fully understand how they are regulated and the molecular mechanisms through which they carry out their action. Uncovering the significance of ncRNAs in the origin of the BAV aortopathy would not only allow a path to find biomarkers for earlier diagnosis, prior to the appearance of symptoms, but also contribute to the development of new RNA-based disease therapies [55].
Additional omic studies in plasma will be essential for the identification of more precise biomarkers that could be used in clinical practice. In addition, basic research with cell and/or animal models should continue in the direction of clarifying the molecular pathways that lead to valve degeneration in order to identify therapeutic targets that are absolutely lacking.
References
- Pedersen, M.W.; Groth, K.A.; Mortensen, K.H.; Brodersen, J.; Gravholt, C.H.; Andersen, N.H. Clinical and pathophysiological aspects of bicuspid aortic valve disease. Cardiol. Young 2019, 29, 1–10.
- Siu, S.C.; Silversides, C.K. Bicuspid aortic valve disease. J. Am. Coll. Cardiol. 2010, 55, 2789–2800.
- Verma, S.; Siu, S.C. Aortic dilatation in patients with bicuspid aortic valve. N. Engl. J. Med. 2014, 370, 1920–1929.
- Michelena, H.I.; Khanna, A.D.; Mahoney, D.; Margaryan, E.; Topilsky, Y.; Suri, R.M.; Eidem, B.; Edwards, W.D.; Sundt, T.M.; Enriquez-Sarano, M. Incidence of Aortic Complications in Patients with Bicuspid Aortic Valves. JAMA 2011, 306, 1104.
- Sorrell, V.L.; Pancyzk, E.; Alpert, J.S. A new disease: Bicuspid aortic valve aortopathy syndrome. Am. J. Med. 2012, 125, 322–323.
- Freeze, S.L.; Landis, B.J.; Ware, S.M.; Helm, B.M. Bicuspid Aortic Valve: A Review with Recommendations for Genetic Counseling. J. Genet. Couns. 2016, 25, 1171–1178.
- Borger, M.A.; Fedak, P.W.M.; Stephens, E.H.; Gleason, T.G.; Girdauskas, E.; Ikonomidis, J.S.; Khoynezhad, A.; Siu, S.C.; Verma, S.; Hope, M.D.; et al. The American Association for Thoracic Surgery consensus guidelines on bicuspid aortic valve-related aortopathy: Executive summary. J. Thorac. Cardiovasc. Surg. 2018, 156, 473–480.
- Michelena, H.I.; Desjardins, V.A.; Avierinos, J.-F.O.; Russo, A.; Nkomo, V.T.; Sundt, T.M.; Pellikka, P.A.; Tajik, A.J.; Enriquez-Sarano, M. Natural History of Asymptomatic Patients with Normally Functioning or Minimally Dysfunctional Bicuspid Aortic Valve in the Community. Circulation 2008, 117, 2776–2784.
- Rodrigues, I.; Agapito, A.F.; de Sousa, L.; Oliveira, J.A.; Branco, L.M.; Galrinho, A.; Abreu, J.; Timoteo, A.T.; Rosa, S.A.; Ferreira, R.C. Bicuspid aortic valve outcomes. Cardiol. Young 2017, 27, 518–529.
- Messner, B.; Bernhard, D. Bicuspid aortic valve-associated aortopathy: Where do we stand? J. Mol. Cell. Cardiol. 2019, 133, 76–85.
- Tzemos, N.; Therrien, J.; Yip, J.; Thanassoulis, G.; Tremblay, S.; Jamorski, M.T.; Webb, G.D.; Siu, S.C. Outcomes in adults with bicuspid aortic valves. JAMA 2008, 300, 1317–1325.
- Della Corte, A.; Bancone, C.; Quarto, C.; Dialetto, G.; Covino, F.E.; Scardone, M.; Caianiello, G.; Cotrufo, M. Predictors of ascending aortic dilatation with bicuspid aortic valve: A wide spectrum of disease expression. Eur. J. Cardiothorac. Surg. 2007, 31, 397–404.
- Martin, P.S.; Kloesel, B.; Norris, R.A.; Lindsay, M.; Milan, D.; Body, S.C. Embryonic Development of the Bicuspid Aortic Valve. J. Cardiovasc. Dev. Dis. 2015, 2, 248–272.
- Elena Sticchi; Rosina De Cario; Alberto Magi; Sabrina Giglio; Aldesia Provenzano; Stefano Nistri; Guglielmina Pepe; Betti Giusti; Bicuspid Aortic Valve: Role of Multiple Gene Variants in Influencing the Clinical Phenotype. BioMed Research International 2018, 2018, 1-9, 10.1155/2018/8386123.
- Stefano Nistri; Maria Cristina Porciani; Monica Attanasio; Rosanna Abbate; Gian Franco Gensini; Guglielmina Pepe; Association of Marfan syndrome and bicuspid aortic valve: Frequency and outcome. International Journal of Cardiology 2012, 155, 324-325, 10.1016/j.ijcard.2011.12.009.
- Carolyn A Bondy; Vladimir K Bakalov; Clara Cheng; Laura Olivieri; Douglas R Rosing; Andrew E Arai; Bicuspid aortic valve and aortic coarctation are linked to deletion of the X chromosome short arm in Turner syndrome. Journal of Medical Genetics 2013, 50, 662-665, 10.1136/jmedgenet-2013-101720.
- R Emanuel; R Withers; K O'brien; P Ross; O Feizi; Congenitally bicuspid aortic valves. Clinicogenetic study of 41 families.. Heart 1978, 40, 1402-1407, 10.1136/hrt.40.12.1402.
- Katrina Huntington; Alasdair G.W Hunter; Kwan-Leung Chan; A Prospective Study to Assess the Frequency of Familial Clustering of Congenital Bicuspid Aortic Valve. Journal of the American College of Cardiology 1997, 30, 1809-1812, 10.1016/s0735-1097(97)00372-0.
- Linda Cripe; Gregor Andelfinger; Lisa J. Martin; Kerry Shooner; D.Woodrow Benson; Bicuspid aortic valve is heritable. Journal of the American College of Cardiology 2004, 44, 138-143, 10.1016/j.jacc.2004.03.050.
- Luis Luna-Zurita; Belén Prados; Joaquim Grego-Bessa; Guillermo Luxán; Gonzalo Del Monte; Alberto Benguría; Ralf H. Adams; José María Pérez Pomares; José Luis De La Pompa; Integration of a Notch-dependent mesenchymal gene program and Bmp2-driven cell invasiveness regulates murine cardiac valve formation. Journal of Clinical Investigation 2010, 120, 3493-3507, 10.1172/jci42666.
- Vidu Garg; Alecia N. Muth; Joshua F. Ransom; Marie K. Schluterman; Robert Barnes; Isabelle N. King; Paul D. Grossfeld; Deepak Srivastava; Mutations in NOTCH1 cause aortic valve disease. Nature 2005, 437, 270-274, 10.1038/nature03940.
- Ruo-Gu Li; Ying-Jia Xu; Juan Wang; Xing-Yuan Liu; Fang Yuan; Ri-Tai Huang; Song Xue; Li Li; Hua Liu; Yan-Jie Li; et al.Xin-Kai QuHong-Yu ShiMin ZhangXing-Biao QiuYi-Qing Yang GATA4 Loss-of-Function Mutation and the Congenitally Bicuspid Aortic Valve. The American Journal of Cardiology 2018, 121, 469-474, 10.1016/j.amjcard.2017.11.012.
- Cristina Alonso-Montes; María Martín; Laura Martínez-Arias; Eliecer Coto; Manuel Naves-Díaz; César Morís; Jorge B. Cannata-Andía; Isabel Rodríguez; Variants in cardiac GATA genes associated with bicuspid aortic valve. European Journal of Clinical Investigation 2018, 48, e13027, 10.1111/eci.13027.
- Brigitte Laforest; Mona Nemer; GATA5 interacts with GATA4 and GATA6 in outflow tract development. Developmental Biology 2011, 358, 368-378, 10.1016/j.ydbio.2011.07.037.
- Ying-Jia Xu; Ruo-Min Di; Qi Qiao; Xiu-Mei Li; Ri-Tai Huang; Song Xue; Xing-Yuan Liu; Juan Wang; Yi-Qing Yang; GATA6 loss-of-function mutation contributes to congenital bicuspid aortic valve. Gene 2018, 663, 115-120, 10.1016/j.gene.2018.04.018.
- Lara Gharibeh; Hiba Komati; Yohan Bossé; Munir Boodhwani; Mahyar Heydarpour; Megan Fortier; Romina Hassanzadeh; Janet Ngu; Patrick Mathieu; Simon Body; et al.Mona NemerOn behalf of the Bicuspid Aortic Valve Consortium GATA6 Regulates Aortic Valve Remodeling, and Its Haploinsufficiency Leads to Right-Left Type Bicuspid Aortic Valve. Circulation 2018, 138, 1025-1038, 10.1161/circulationaha.117.029506.
- Wei-Feng Jiang; Ying-Jia Xu; Cui-Mei Zhao; Xin-Hua Wang; Xing-Biao Qiu; Xu Liu; Shao-Hui Wu; Yi-Qing Yang; A novel TBX5 mutation predisposes to familial cardiac septal defects and atrial fibrillation as well as bicuspid aortic valve. Genetics and Molecular Biology 2020, 43, e20200142, 10.1590/1678-4685-gmb-2020-0142.
- Elisabeth Gillis; Ajay A. Kumar; Ilse Luyckx; Christoph Preuss; Elyssa Cannaerts; Gerarda Van De Beek; Björn Wieschendorf; Maaike Alaerts; Nikhita Bolar; Geert Vandeweyer; et al.Josephina MeesterFlorian WünnemannRussell A. GouldRustam ZhurayevDmytro ZerbinoSalah A. MohamedSeema MitalLuc MertensHanna M. BjörckAnders Franco-CerecedaAndrew S. McCallionLut Van LaerJudith M. A. VerhagenIngrid M. B. H. Van De LaarMarja W. WesselsEmmanuel MessasGuillaume GoudotMichaela NemcikovaAlice KrebsovaMarlies KempersSimone SaleminkToon DuijnhouwerXavier JeunemaitreJuliette AlbuissonPer ErikssonGregor AndelfingerHarry C. DietzAline VerstraetenBart L. Loeys Candidate Gene Resequencing in a Large Bicuspid Aortic Valve-Associated Thoracic Aortic Aneurysm Cohort: SMAD6 as an Important Contributor. Frontiers in Physiology 2017, 8, 400, 10.3389/fphys.2017.00400.
- Ilse Luyckx; Gretchen MacCarrick; Marlies Kempers; Josephina Meester; Céline Geryl; Olivier Rombouts; Nils Peeters; Charlotte Claes; Nele Boeckx; Natzi Sakalihasan; et al.Adeline JacquinetAlexander HoischenGeert VandeweyerSarah Van LentJohan SaenenEmeline Van CraenenbroeckJanneke TimmermansAnthonie DuijnhouwerHarry DietzLut Van LaerBart LoeysAline Verstraeten Confirmation of the role of pathogenic SMAD6 variants in bicuspid aortic valve-related aortopathy. European Journal of Human Genetics 2019, 27, 1044-1053, 10.1038/s41431-019-0363-z.
- M Attanasio; I Lapini; L Evangelisti; L Lucarini; Betti Giusti; Mc Porciani; R Fattori; C Anichini; R Abbate; Gf Gensini; et al.G Pepe FBN1 mutation screening of patients with Marfan syndrome and related disorders: detection of 46 novel FBN1 mutations. Clinical Genetics 2008, 74, 39-46, 10.1111/j.1399-0004.2008.01007.x.
- Russell A. Gould; Baylor-Hopkins Center For Mendelian Genomics; Hamza Aziz; Courtney E. Woods; Manuel Alejandro Seman-Senderos; Elizabeth Sparks; Christoph Preuss; Florian Wünnemann; Djahida Bedja; Cassandra R. Moats; et al.Sarah A. McClymontRebecca RoseNara SobreiraHua LingGretchen MacCarrickAjay Anand KumarIlse LuyckxElyssa CannaertsAline VerstraetenHanna M. BjörkAnn-Cathrin LehsauVinod Jaskula-RangaHenrik LauridsenAsad A. ShahChristopher L. BennettPatrick T. EllinorHonghuang LinEric M. IsselbacherChristian Lacks Lino CardenasJonathan ButcherG. Chad HughesMark E. LindsayLuc MertensAnders Franco-CerecedaJudith M. A. VerhagenMarja WesselsSalah A. MohamedPer ErikssonSeema MitalLut Van LaerBart L. LoeysGregor AndelfingerAndrew S. McCallionHarry C. DietzMIBAVA Leducq Consortium ROBO4 variants predispose individuals to bicuspid aortic valve and thoracic aortic aneurysm. Nature Genetics 2018, 51, 42-50, 10.1038/s41588-018-0265-y.
- Dong-Chuan Guo; Hariyadarshi Pannu; Van Tran-Fadulu; Christina L Papke; Robert K Yu; Nili Avidan; Scott Bourgeois; Anthony L Estrera; Hazim J Safi; Elizabeth Sparks; et al.David J AmorLesley AdesVivienne McConnellColin WilloughbyDianne N AbueloMarcia C WillingRichard A LewisDong KimSteve SchererPoyee P TungChul AhnLouis Maximilian BujaC. S. RamanSanjay S SheteDianna M Milewicz Mutations in smooth muscle α-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nature Genetics 2007, 39, 1488-1493, 10.1038/ng.2007.6.
- Luca Longobardo; Maria Ludovica Carerj; Gabriele Pizzino; Alessandra Bitto; Maurizio Cusma Piccione; Marta Zucco; Lilia Oreto; Maria Chiara Todaro; Maria Pia Calabrò; Francesco Squadrito; et al.Gianluca Di BellaGiuseppe OretoBijoy K. KhandheriaScipione CarerjConcetta Zito Impairment of elastic properties of the aorta in bicuspid aortic valve: relationship between biomolecular and aortic strain patterns. European Heart Journal - Cardiovascular Imaging 2017, 19, 879-887, 10.1093/ehjci/jex224.
- Radoslaw Debiec; Stephen E. Hamby; Peter D. Jones; Sue Coolman; Manish Asiani; Shireen Kharodia; Gregory J. Skinner; Nilesh J. Samani; Tom R. Webb; Aidan Bolger; et al. Novel loss of function mutation in NOTCH1 in a family with bicuspid aortic valve, ventricular septal defect, thoracic aortic aneurysm, and aortic valve stenosis. Molecular Genetics & Genomic Medicine 2020, 8, e1437, 10.1002/mgg3.1437.
- Wilhelmina S. Kerstjens-Frederikse; Ingrid M. B. H. Van De Laar; Yvonne J. Vos; Judith M. A. Verhagen; Rolf M. F. Berger; Klaske D. Lichtenbelt; Jolien S. Klein Wassink-Ruiter; Paul A. Van Der Zwaag; Gideon J. Du Marchie Sarvaas; Klasien A. Bergman; et al.Catia M. BilardoJolien W. Roos-HesselinkJohan H. P. JanssenIngrid M. Frohn-MulderKarin Y. Van Spaendonck-ZwartsJoost P. Van MelleRobert M. W. HofstraM. W. Wessels Cardiovascular malformations caused by NOTCH1 mutations do not keep left: data on 428 probands with left-sided CHD and their families. Genetics in Medicine 2016, 18, 914-923, 10.1038/gim.2015.193.
- Jamieson Whitcomb; Lara Gharibeh; Mona Nemer; From embryogenesis to adulthood: Critical role for GATA factors in heart development and function. IUBMB Life 2019, 72, 53-67, 10.1002/iub.2163.
- Gianluca Lorenzo Perrucci; Erica Rurali; Aoife Gowran; Alessandro Pini; Carlo Antona; Roberto Chiesa; Giulio Pompilio; Patrizia Nigro; Vascular smooth muscle cells in Marfan syndrome aneurysm: the broken bricks in the aortic wall. Cellular and Molecular Life Sciences 2016, 74, 267-277, 10.1007/s00018-016-2324-9.
- Guglielmina Pepe; Stefano Nistri; Betti Giusti; Elena Sticchi; Monica Attanasio; Cristina Porciani; Rosanna Abbate; Robert O Bonow; Magdi Yacoub; Gian Franco Gensini; et al. Identification of fibrillin 1 gene mutations in patients with bicuspid aortic valve (BAV) without Marfan syndrome. BMC Medical Genetics 2014, 15, 23-23, 10.1186/1471-2350-15-23.
- Giada Tortora; Anita Wischmeijer; Paolo Berretta; Jacopo Alfonsi; Luca Di Marco; Andrea Barbieri; Caterina Marconi; Federica Isidori; Cesare Rossi; Ornella Leone; et al.Roberto Di BartolomeoMarco SeriDavide Pacini Search for genetic factors in bicuspid aortic valve disease: ACTA2 mutations do not play a major role. Interactive Cardiovascular and Thoracic Surgery 2017, 25, 813-817, 10.1093/icvts/ivx242.
- Barbara Messner; David Bernhard; Bicuspid aortic valve-associated aortopathy: Where do we stand?. Journal of Molecular and Cellular Cardiology 2019, 133, 76-85, 10.1016/j.yjmcc.2019.05.023.
- Hope, M.D.; Hope, T.A.; Crook, S.E.; Ordovas, K.G.; Urbania, T.H.; Alley, M.T.; Higgins, C.B. 4D flow CMR in assessment of valve-related ascending aortic disease. JACC Cardiovasc. Imaging 2011, 4, 781–787.
- Mahadevia, R.; Barker, A.J.; Schnell, S.; Entezari, P.; Kansal, P.; Fedak, P.W.; Malaisrie, S.C.; McCarthy, P.; Collins, J.; Carr, J.; et al. Bicuspid aortic cusp fusion morphology alters aortic three-dimensional outflow patterns, wall shear stress, and expression of aortopathy. Circulation 2014, 129, 673–682.
- Ruzmetov, M.; Shah, J.J.; Fortuna, R.S.; Welke, K.F. The Association Between Aortic Valve Leaflet Morphology and Patterns of Aortic Dilation in Patients with Bicuspid Aortic Valves. Ann. Thorac. Surg. 2015, 99, 2101–2107.
- Jackson, V.; Petrini, J.; Caidahl, K.; Eriksson, M.J.; Liska, J.; Eriksson, P.; Franco-Cereceda, A. Bicuspid aortic valve leaflet morphology in relation to aortic root morphology: A study of 300 patients undergoing open-heart surgery. Eur. J. Cardiothorac. Surg. 2011, 40, e118–e124.
- Habchi, K.M.; Ashikhmina, E.; Vieira, V.M.; Shahram, J.T.; Isselbacher, E.M.; Sundt, T.M., 3rd; Shekar, P.; Muehlschlegel, J.D.; Bicuspid Aortic Valve, C.; Body, S.C. Association between bicuspid aortic valve morphotype and regional dilatation of the aortic root and trunk. Int. J. Cardiovasc. Imaging 2017, 33, 341–349.
- Bissell, M.M.; Hess, A.T.; Biasiolli, L.; Glaze, S.J.; Loudon, M.; Pitcher, A.; Davis, A.; Prendergast, B.; Markl, M.; Barker, A.J.; et al. Aortic dilation in bicuspid aortic valve disease: Flow pattern is a major contributor and differs with valve fusion type. Circ. Cardiovasc. Imaging 2013, 6, 499–507.
- Guzzardi, D.G.; Barker, A.J.; van Ooij, P.; Malaisrie, S.C.; Puthumana, J.J.; Belke, D.D.; Mewhort, H.E.; Svystonyuk, D.A.; Kang, S.; Verma, S.; et al. Valve-Related Hemodynamics Mediate Human Bicuspid Aortopathy: Insights from Wall Shear Stress Mapping. J. Am. Coll. Cardiol. 2015, 66, 892–900.
- Fedak, P.W.; de Sa, M.P.; Verma, S.; Nili, N.; Kazemian, P.; Butany, J.; Strauss, B.H.; Weisel, R.D.; David, T.E. Vascular matrix remodeling in patients with bicuspid aortic valve malformations: Implications for aortic dilatation. J. Thorac. Cardiovasc. Surg. 2003, 126, 797–806.
- Shan, Y.; Li, J.; Wang, Y.; Wu, B.; Barker, A.J.; Markl, M.; Wang, C.; Wang, X.; Shu, X. Aortic shear stress in patients with bicuspid aortic valve with stenosis and insufficiency. J. Thorac. Cardiovasc. Surg. 2017, 153, 1263–1272.e1.
- Bollache, E.; Guzzardi, D.G.; Sattari, S.; Olsen, K.E.; Di Martino, E.S.; Malaisrie, S.C.; van Ooij, P.; Collins, J.; Carr, J.; McCarthy, P.M.; et al. Aortic valve-mediated wall shear stress is heterogeneous and predicts regional aortic elastic fiber thinning in bicuspid aortic valve-associated aortopathy. J. Thorac. Cardiovasc. Surg. 2018, 156, 2112–2120.e2.
- Della Corte, A.; Bancone, C.; Dialetto, G.; Covino, F.E.; Manduca, S.; Montibello, M.V.; De Feo, M.; Buonocore, M.; Nappi, G. The ascending aorta with bicuspid aortic valve: A phenotypic classification with potential prognostic significance. Eur. J. Cardiothorac. Surg. 2014, 46, 240–247.
- Galian-Gay, L.; Carro Hevia, A.; Teixido-Tura, G.; Rodriguez Palomares, J.; Gutierrez-Moreno, L.; Maldonado, G.; Gonzalez-Alujas, M.T.; Sao-Aviles, A.; Gallego, P.; Calvo-Iglesias, F.; et al. Familial clustering of bicuspid aortic valve and its relationship with aortic dilation in first-degree relatives. Heart 2019, 105, 603–608.
- Girdauskas, E.; Rouman, M.; Disha, K.; Espinoza, A.; Misfeld, M.; Borger, M.A.; Kuntze, T. Aortic Dissection After Previous Aortic Valve Replacement for Bicuspid Aortic Valve Disease. J. Am. Coll. Cardiol. 2015, 66, 1409–1411.
- Fatehi Hassanabad, A.; Garcia, J.; Verma, S.; White, J.A.; Fedak, P.W.M. Utilizing wall shear stress as a clinical biomarker for bicuspid valve-associated aortopathy. Curr. Opin. Cardiol. 2019, 34, 124–131.
- Pulignani, S.; Borghini, A.; Andreassi, M.G. microRNAs in bicuspid aortic valve associated aortopathy: Recent advances and future perspectives. J. Cardiol. 2019, 74, 297–303.


