Homocysteine thiolactone is a five-membered cyclic thioester of amino acid homocysteine. It is generated from homocysteine as a result of an error-editing reaction, principally, of methionyl-tRNA synthetase. An elevated level of homocysteine thiolactone is associated with cardiovascular diseases, strokes, atherosclerosis, neurological abnormalities, etc., presumably because it reacts to the side chain of protein lysine causing protein damage and autoimmune responses. It is not only an important metabolite but also a versatile building block for organic and bioorganic synthesis. This entry contains data on the homocysteine thiolactone formation, metabolism, toxicity mechanism in vivo, and the bioorganic chemistry applications as a powerful synthetic tool in polymer science, sustainable materials development, and probes.
Homocysteine (Hcy) is a non-protein amino acid that is an important risk factor for arteriosclerosis, Alzheimer’s disease, cardiovascular disease, ischemic heart disease, stroke, cancer, diabetic retinopathy, and diseases of the central nervous system in humans
[1][2][3][4][5][6][7][8][9][10][11][12]. More than one hundred conditions are associated with raised concentrations of Hcy
[7][10][13][14]. Following Hcy discovery in 1932 by Vincent du Vigneaud, Hcy was characterized as an important intermediate in methionine metabolism. However, only after 40 years, the elevated level of Hcy in the blood (hyperhomocysteinemia) formed in a pioneer theory, Hcy theory of arteriosclerosis and cardiovascular diseases, by Kilmer S. McCully
[1][2][15][16][17]. He noted that thromboembolic disease was a characteristic feature of the inborn errors pointing to Hcy as a risk factor. The theory implicates hyperhomocysteinemia along with cholesterol as the key factors in the production of vascular disease and arteriosclerosis in the general population.
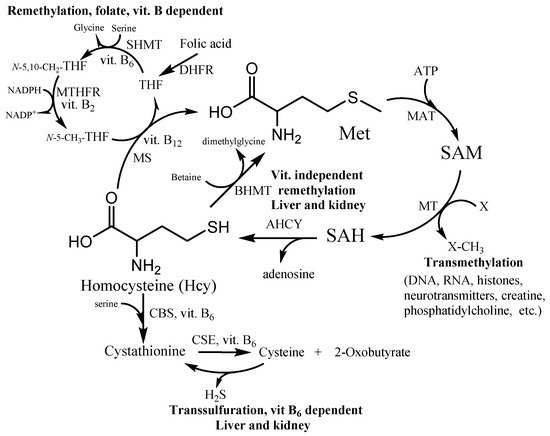
Hcy takes part in many fundamental processes in the human organism. It is formed in the methionine cycle as an intermediate by hydrolysis of S-adenosylhomocysteine to Hcy and adenosine (Figure 1). Under normal conditions, approximately 50% of Hcy is remethylated to form methionine. Two distinct routes exist for remethylation. The first reaction is the vitamins-dependent remethylation by methionine synthase (MS). The N-5-methyl tetrahydrofolate can donate a methyl group to Hcy in a reaction catalyzed by the vitamin B12-dependent enzyme. The folate cycle also requires folic acid, vitamin B2 and B6, and NADPH. The second reaction uses betaine as a methyl group donor for methionine synthesis by enzyme betaine-homocysteine methyltransferase (BHMT). The reaction presumably occurs in the liver, kidney, and lens, whereas the first vitamin-dependent route is found in all tissues. The other possibility of Hcy irreversibly consumption is the transsulfuration pathway. The first reaction is a condensation between Hcy and serine (Ser) leading to cystathionine production by cystathionine β-synthase (CBS). The cystathionine is further hydrolyzed to Cys and α-ketobutyrate by cystathionine γ-lyase (CSE). These two reactions are catalyzed by the vitamin B6-dependent enzymes (CSE and CBS). The Hcy detoxification through the transsulfuration pathway occurs in the liver, kidney, small intestine, and lens. Thus, the vitamins B and folate status play an important role in Hcy and its metabolites balance within the cell and subsequently the plasma level circulation.
Figure 1. Homocysteine (Hcy) metabolic pathway. SAM—S-adenosylmethionine; SAH—S-adenosylhomocysteine; MS—methionine synthase; BHMT—betaine-homocysteine methyltransferase; MT—SAM-dependent methyltransferases; MAT—methionine adenosyltransferase; AHCY—S-adenosylhomocysteine hydrolase; CBS—cystathionine β-synthase; CSE—cystathionine γ-lyase; THF—tetrahydrofolate; N-5-CH3-THF—5-methyltetrahydrofolate; N-5,10-CH2-THF—N5,N10-methylenetetrahydrofolate; DHFR—dihydrofolate reductase; SHMT—serinehydroxymethyltransferase; MTHFR—methylentetrahydrofolate reductase.
One option to obtain an elevated level (as high as 400–500 µmol/L) of Hcy is the mutations in the genes coding enzymes involved in Hcy or related metabolisms such as CBS, MS, BHMT, and MTHFR
[18]. Vitamin B and folic acid deficiencies and an imbalanced diet may also lead to hyperhomocysteinemia
[11][19]. Clinical trials show strong associations between low levels of folate, vitamin B
12, and Hcy serum concentrations and various pathologies
[11][20][21]. The most common among them are Alzheimer’s disease, Parkinson’s disease, autism, schizophrenia, bipolar disorder, vascular dementia, peripheral neuritis, and stroke. Therapy with multiple vitamins and folate might be one key to correct Hcy level
[11][18][19][21], helping in several pathological conditions and also in pregnancy. However, different strategies to reduce plasma Hcy concentrations have reached inconsistent results
[10][11][21]. For example, the same folic acid dose decreases the concentration of Hcy by 25–45%
[11]. In most cases, lab results are more encouraging than clinical trials
[21]. The failure of Hcy-lowering by folic acid and vitamins B
6 and B
12 by clinical trials to reduce heart attacks and cardiovascular mortality, suggests that some changes are irreversible and highlights the need for additional research into the mechanisms by which Hcy metabolites cause the disease
[22][23].
Genetic abnormalities and vitamin deficiencies explain only a part of Hcy-associated pathologies
[12]. One of the explanations is that Hcy metabolism interacts with complex biochemical pathways involving the cooperation of multiple enzymes and producing various molecules that are essential biochemical steps for cell survival. For example, Hcy increases tau protein damage via several possible mechanisms
[21][24]: cyclin-dependent kinase 5 (cdk5) activation and inactivation of protein phosphatase 2A (PP2A); 20S proteasome inactivation; caspase3 activation and increased C-terminal truncated tau by caspase. Tau is a microtubule-associated protein present in the cytoskeleton of neuronal cells and helps maintain neuronal cell integrity. This process leads to tau phosphorylation, truncation, oligomerization, and toxic aggregates formation
[24]. Despite the widespread involvement of Hcy in these pathways, it remains relatively unclear exactly how Hcy leads to the formation of toxic tau species and neuronal death.
Many metabolic, hormonal, and epigenetic factors interact with Hcy metabolism. One of the most important examples is reduced synthesis of SAM. SAM is used as a universal methyl donor not only in a variety of biosynthesis of different compounds (phospholipids, proteins, nucleic acids, etc.) but also in epigenetic modulations (
Figure 1)
[22]. Therefore, the Hcy level affects epigenetic regulation and leads to alterations in gene expression via DNA methylation, histone modification, changing chromatin structure, and the action of non-coding RNA
[22][23][25][26]. How Hcy affects gene expression in experimental animals depends on many factors, including diet, mutations, and vitamin status. Dysregulation in methylation status is among the central mechanisms that explain the negative effect of high Hcy levels related to vascular diseases and brain disorders. Moreover, hyperhomocysteinemia disrupts the blood-brain barrier in humans which can lead to various pathologies
[9].
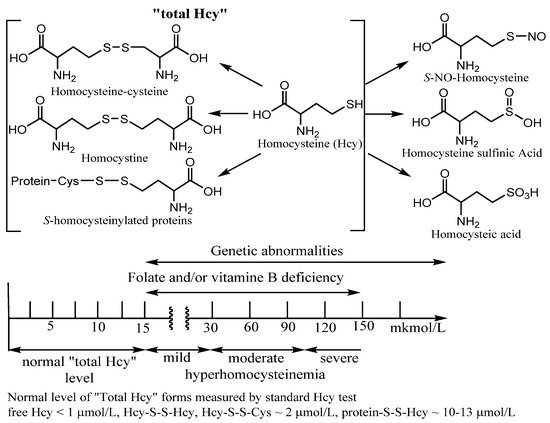
Approximately 10% of the total daily cellular production of Hcy that is not metabolized within the cell is exported to the plasma. Under normal conditions, the total plasma content of Hcy varies from 3–5 to 15 µM. An elevated plasma level of Hcy is known as hyperhomocysteinemia (in urea homocystinuria). The ranges of Hcy elevated levels in plasma have been referred to as mild (16–30 µM), moderate (31–100 µM), or severe (>100 µM). Moderate hyperhomocysteinemia was found in subjects with impaired renal function and with end-stage renal disease. Patients with genetic abnormalities and inborn errors of Hcy metabolism usually have severe hyperhomocysteinemia (
Figure 2)
[5].
Figure 2. Examples of Homocysteine products of metabolism. “Total homocysteine” level in plasma in healthy patients and under vitamin deficiency and genetic disorders. Many Hcy metabolism products as homocysteic acid, homocysteine sulfinic acid, homocysteine, and S-homocysteinylated proteins are beyond this entry.
When measuring “total Hcy”, it is important to measure all forms of Hcy to get true results of the Hcy status. However, only four forms of Hcy make up the “total Hcy” using a standard commercial Hcy kit. Among them are homocysteine (Hcy), Hcy mixed disulfides, and
S-homocysteinylated proteins (
Figure 2). Using the standard kit procedure, a reducing reagent is added to the sample to form free Hcy amino acid from the latter three metabolites with straightforward Hcy identification by various methods. Under physiological conditions, less than 1% of total Hcy is present in a free reduced form. About 15% has been found in different oxidized forms (Hcy-S-S-Hcy, Hcy-S-S-Cys). However, the majority of plasma total Hcy (~80%) is
S-homocysteinylated and
N-homocysteinylated proteins. The precursor of the main fraction as
N-homocysteinylated proteins is cyclic thioester, homocysteine thiolactone (HTL) (
Figure 3). Because of the structural similarity to methionine, Hcy can be recognized and activated by methionyl-tRNA synthetase (and some other aminoacyl-tRNA synthetases
[27][28]). It can form activated derivative Hcy~AMP and then catalyzes an intramolecular thioester bond formation in a reaction between the side-chain—SH group and activated carbonyl group of Hcy, affording HTL (
Figure 3). Hcy editing prevents its access to the genetic code and occurs at the synthetic/editing catalytic module of the enzyme which contains a thiol-binding site. Hcy is not transferred to tRNA, and it is not incorporated into protein. All organisms edit Hcy by the conversion into HTL
[28][29]. The energy of the anhydride/macroergic bond of Hcy~AMP is conserved in the thioester bond of HTL.
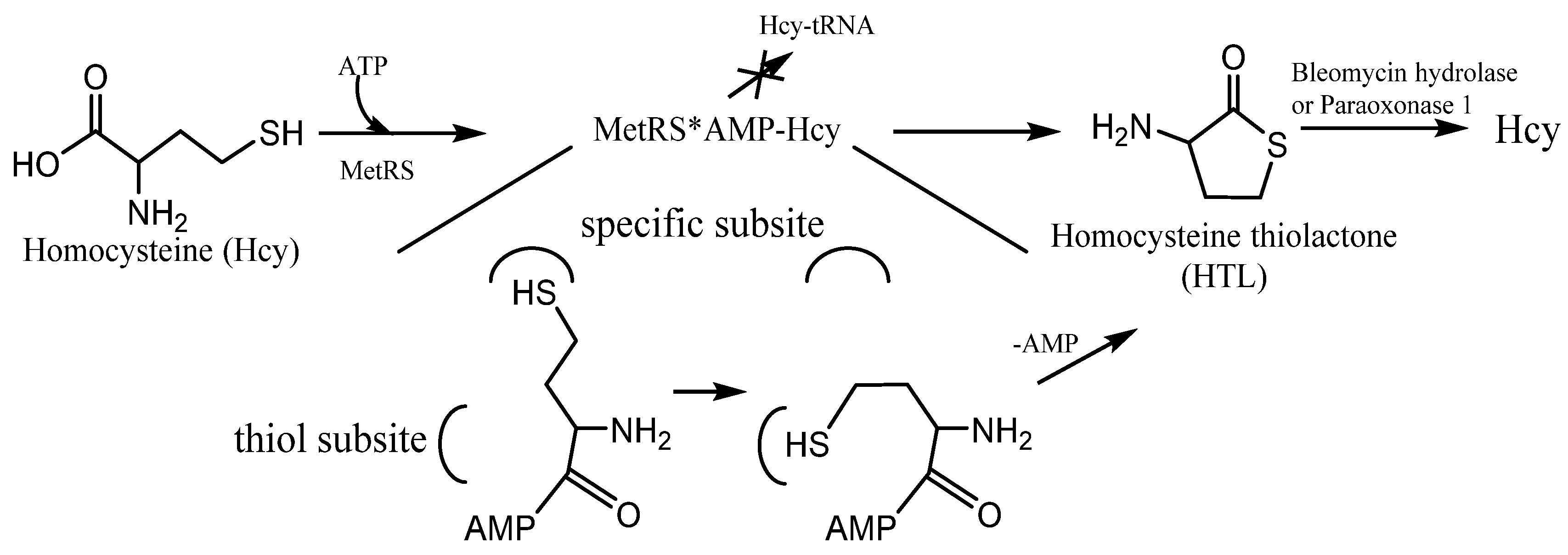
Figure 3. The mechanism of Homocysteine thiolactone (HTL) formation: editing of Hcy at the active site of aminoacyl-tRNA synthetases. MetRS—methionine-tRNA synthetase. MetRS*AMP-Hcy—MetRS and AMP-Hcy complex.
For many years and to date, many studies are undertaken to understand Hcy metabolism and the mechanisms of its toxicity. Several main pathways of Hcy toxicity have been discussed in the literature: (1) protein modification (
S- and
N-homocysteinylation); (2) oxidative stress induction; and (3) excitotoxicity (nerve cells suffer damage or death). There are many Hcy metabolites such as homocysteic acid, homocysteine sulfinic acid, homocysteine, and
S-homocysteinylated proteins that are beyond this entry (
Figure 2)
[30]. This entry is intended to be a summary of several pieces of evidence of the homocysteine thiolactone (HTL) theory of Hcy toxicity. Several studies
[5][30][31][32][33][34][35][36][37] have found that HTL elevated level is associated with pathological conditions such as atherosclerosis and neurodegenerative diseases, vascular damage in diabetic patients, and cardiovascular disease. Due to the high reactivity of HTL, its discovery in living cells and the Hcy theory of arteriosclerosis and cardiovascular diseases lead to intensive investigations of its possible reactions with proteins (
Figure 4).
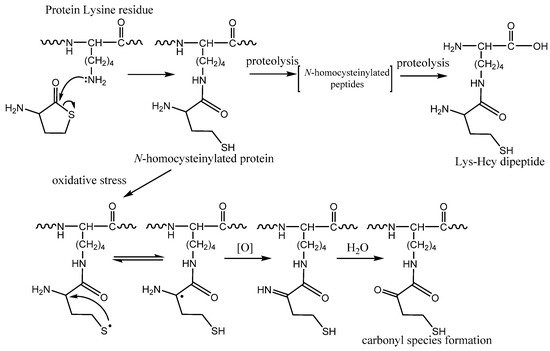
Figure 4. Protein N-homocysteinylation: chemical modification of a lysine residue by homocysteine thiolactone (HTL). Formation of dipeptide Nε-Hcy-Lys by protein proteolysis. The mechanism of carbonyl species formation by N-homocysteinylated proteins caused by oxidative stress.
HTL is a stable thioester in water solution (pH = 7.4, a half-life of ~24–30 h)
[33][34], with facile reactions at its amino and activated-carboxyl group, respectively. As electrophile, it reacts with several types of nucleophiles in vivo: amines (protein lysine side chains,
Figure 4,
N-homocysteinylation) and hydroxyl or water (hydrolysis). The amino group of HTL reacts with the aldehyde group of pyridoxal phosphate (vitamin B
6) forming tetrahydrothiazine
[38]. Similar to the condensation of HTL with pyridoxal phosphate, Hcy reacts with pyridoxal phosphate yielding tetrahydrothiazine
[38]. The facile formation of stable tetrahydrothiazines raises one more possible mechanisms of inactivation of pyridoxal phosphate by Hcy and HTL in vivo. On the other way, the facile formation of metabolically inactive tetrahydrothiazine may contribute to the elimination of harmful high reactive metabolites (HTL) from the human body.
Similar to other harmful products of normal metabolism, HTL is also eliminated by urinary excretion
[30][31]. HTL concentrations in urine vary from 10 nM to 500 nM (3–30% of total urinary Hcy) and are 100-fold higher than in plasma
[30]. Renal excretion removes a large amount of HTL that would otherwise cause protein
N-homocysteinylation. The remainder of the HTL is metabolized by two major pathways: (1) enzymatic hydrolysis to Hcy (bleomycin hydrolase, paraoxonase 1,
Figure 3 [39]); (2) reaction with the side chain of protein lysine forming
N-homocysteinylated proteins (
Figure 4). Bleomycin hydrolase and paraoxonase 1 protect the organism against neurotoxicity induced by HTL. However, under organism disorder presented above, kidney disorders, and chronic hyperhomocysteinemia, there is an elevated level of HTL and
N-homocysteinylated proteins. In healthy humans, plasma HTL concentration is usually ranged from one to five nM (sometimes to 35 nM) and increases when remethylation or transsulfuration reactions are impaired by genetic alterations of enzymes, vitamin deficiency, or other metabolism disorders
[30][31][32][37]. When methionine synthase activity is inhibited by folate or vitamin B
12 deprivation, almost all Hcy converts into HTL. Plasma HTL concentration is elevated in humans with MTHFR (methylenetetrahydrofolate reductase) and CBS deficiency, and can be up to 7 mM and, rare, to the 50 mM in patients with coronary artery disease
[30][31][32].
HTL is much more toxic than Hcy and induces cell apoptosis at low concentrations
[31]. The extent of apoptosis in HUVEC cells treated with 200 μM HTL and Hcy corresponds to 30% and 1% of apoptotic cells, respectively. Treatments with HTL are known to cause toxicity in experimental animals
[30][31]. For example, an animal diet supplemented with HTL causes lethality, growth retardation, developmental abnormalities, atherosclerosis, and arterial thrombosis.
[30]. Previous studies on the effect of L- and D-stereoisomers of HTL have demonstrated the same compound toxicity to rat embryos. In contrast, L-Hcy is toxic to rat embryos, whereas D-Hcy is not
[33]. The lack of toxicity of D-Hcy is consistent with the stereoselectivity of methionyl-tRNA synthetase, whose active site binds only L-Hcy and produces L-HTL. A possible explanation for this might be that both stereoisomers of HTL can chemically react with protein Lys residues. In addition, HTL can easily diffuse through the cell membrane (and maybe also nucleus) due to the most molecules’ neutral charge in vivo because of the relatively low amino group pK
a value (~7.1)
[40]. Therefore, HTL can easily modify plasma proteins leading to different disorders.
The protein
N-homocysteinylation process usually occurs on hyper-reactive lysine species in proteins which prevent natural protein lysine residues modification. It can affect epigenetic regulation of gene expression (prevents acetylation and methylation of histone Lys residues) and biogenesis of collagen (prevents pyridinoline cross-link formation)
[31]. DNA and histone modifications have been identified to have a crucial role in the progression of many disorders/diseases such as atherosclerosis, stroke, Alzheimer’s disease, and cancer
[22]. Hcy has a wide range of biological effects and pathophysiological changes associated with its possible metabolites. As mentioned above, the vitamin efficiency clinical trials suggest that some diseases are associated with some irreversible changes, and cannot be explained by the simple level of total Hcy. Moreover, total Hcy is a four-component marker (
Figure 2), which nevertheless does not include HTL, SAM, and
N-homocysteinylated proteins. The changes in gene expression caused by Hcy metabolites allow in identifying biological pathways and human diseases
[41]. It was found that genes affected by HTL,
N-homocysteinylated proteins, and Hcy were significantly enriched in 30, 13, and seven molecular pathways, respectively. Only five common pathways were enriched in genes affected by all three metabolites: lipid, fatty acid, and steroid metabolism; blood coagulation; wound healing; cysteine and methionine metabolism; sulfur amino acid biosynthesis. These findings suggest that toxic Hcy metabolites up-regulate the expression of genes involved in the removal of Hcy excess
[41].
N-homocysteinylated proteins and Hcy did not overlap the majority of HTL-affected pathways. Among the top-five HTL pathways are chromatin organization, one-carbon metabolism, lipid localization, lipoprotein metabolic processes, and lipid and fatty acid transport. Moreover, chromatin organization was the top molecular pathway affected by the HTL level
[41]. Notably, the histone-related genes were not influenced by Hcy or
N-homocysteinylated proteins.
N-homocysteinylated proteins affected six diverse pathways: organic acid biosynthetic process, focal adhesion, biological oxidations, endothelial cell differentiation, epithelial cell differentiation, and plasma lipoprotein particle remodeling. Two pathways as aspartate family amino acid metabolic process and seleno amino acid metabolism were associated with Hcy
[41]. Presented results suggest that HTL and
N-homocysteinylated proteins, rather than Hcy, cause changes in gene expression for the majority of genes and biological pathways. According to this data
[41], a specific Hcy metabolite is mostly associated with a specific disease (
Table 1).
Table 1. Diseases and disorders are mostly associated with Hcy metabolites according to work
[41].
| Hcy Metabolite |
Disease and Disorders |
| Hcy, HTL, and N-homocysteinylated proteins |
Cancer, cerebrovascular disease, ischemic disease, atherosclerosis, coronary and congenital heart diseases, Alzheimer’s disease, and neural tube defects |
| Hcy |
Amino acid and lipid metabolism |
| HTL |
Cardiovascular disease, myocardial infarction, dyslipidemia, deep vein thrombosis, venous thromboembolism, stroke, cardiac infarction, skeletal, and muscular systems |
| N-homocysteinylated proteins |
Cardiovascular disease, metabolic disease, neurological disease, dermatological disease, and placental abruption |
N-homocysteinylated proteins are a significant component of Hcy metabolism in humans. The major
N-homocysteinylation targets include human hemoglobin, human serum albumin (HSA), and γ–globulin. Among other targets are antitrypsin, transferrin, fibrinogen, low- and high-density lipoproteins (LDL and HDL, respectively), cytochrome c, and histones (
Table 2). Despite
N-homocysteinylated proteins represent ~0.5–13 μM linked Hcy in healthy human plasma (the results highly depend on the quantification method and patients’ conditions
[31][34][40]), this process impairs or alters the protein’s function, proteolysis stability, oligomerization possibility, aggregation, amyloid transformation, etc.
[30][31][32] (
Table 2). Protein
N-homocysteinylation leads to the formation of a new thiol group (
Figure 4), which influences protein’s susceptibility to oxidation, increases intramolecular disulfide bond formation, and multimerization. Taking into account the fact that in humans about 70–80% of circulating Hcy is linked to blood proteins, the
N-homocysteinylated protein aggregates can be extremely toxic and should be considered as a risk factor of conformational diseases.
Table 2 presents some examples of the harmful influence of HTL modification on protein functions. For example, HSA with mostly
α-helical secondary structure undergoes
N-homocysteinylation, loses helical content, forms oligomers, and converts to amyloid-like
β-sheet structures
[31][42]. Recently
[43], it was shown that HSA forms reversible dimers in water solution.
N-homocysteinylation of HSA influences the dimerization process, accelerates it to various much stable structures. Dimer formation seems to have allosteric effects on HSA and could influence the many physiological functions of HSA. Considering that HSA is the most abundant protein and an important hub where many physiological processes, pathologies, and therapeutics intersect,
N-homocysteinylation leads to unimaginable consequences that require additional study. Moreover,
N-homocysteinylation decrease the proteolytic degradation efficiency of HSA by trypsin and chymotrypsin
[44]. Combined with a high half-life
N-homocysteinylated HSA time (~20 days) and the possibility to form free radicals
[45], the protein can be extremely toxic to the organism.
Table 2. Protein
N-homocysteinylation: examples and functional consequences
[5][30][31][46].
| Protein |
Function |
Effect of N-Homocysteinylation |
| Hemoglobin Myoglobin |
Oxygen transport |
Oxidative damage, aggregation |
| Albumin |
Transport of metabolic products, fatty acid, etc.; maintenance of the blood within the vascular system |
Structural changes, influence the function of Cys-34, susceptibility to proteolysis, binding properties, increase oligomer formation, amyloidal transformation |
| γ–globulin |
Antibodies |
Aggregation |
| Fibrinogen |
Precursor of fibrin |
Aggregation, fibers thinner and more resistant to fibrinolysis |
| Trypsin |
Cleavage of amide bonds of Arg and Lys |
Partly or complete inactivation, complete inactivation when ~88% Lys residues modified |
| Cytochrome c |
Electron transport |
Oxidative damage, aggregation |
| RNase A |
RNA hydrolysis |
Aggregation |
| Crystallin |
Eye structural protein |
Aggregation |
| Histones |
Create structural units with DNA, DNA packing |
Prevent epigenetic regulation, changes in gene expression |
| Methionyl-tRNA synthetase |
Aminoacylation of tRNAMet with methionine |
Complete inactivation when ~33% Lys residues modified |
| Tau |
Stabilization of neuronal microtubules |
Altered tubulin-binding resulting in enhances self-association and aggregation |
| Prion |
Cell signaling, neuritogenesis, neuronal homeostasis |
Aggregation and amyloid transformation |
Some pathology associated with hyperhomocysteinemia is also linked to oxidative stress. Aberrant Hcy metabolism leads to redox imbalance according to two possible mechanisms. Homocysteinylation of proteins affects the function and activity of different enzymes, such as superoxide dismutase, catalase, or glutathione peroxidase. By itself,
N-homocysteinylation leads to the formation of thiols groups which change the redox status of the protein. The presented process can increase the formation of reactive oxygen and nitrogen species, followed by biopolymers oxidation. Oxidative damage caused by
N-homocysteinylation was demonstrated for HSA, hemoglobin, myoglobin, and cytochrome c
[31]. Recently
[45], it was found that Hcy residue in protein under physiological conditions can form thiyl radical (
Figure 4). Interestingly, it is possible kinetically favored intramolecular hydrogen atom transfer and formation of αC radicals promote carbonyl formation, multiple fragmentation products, and protein cross-links
[45]. Another confirmation of the importance of the processes is that the blocking of the α-amino group of HTL inhibits the αC radicals formation and HSA oligomerization
[47]. For HSA, major plasma protein, αC radicals formation in vitro occurs at physiological temperature, 37 °C, without the addition of active oxygen forms or free-radical initiator systems
[45]. The same αC radical formation was also found for the
Nε-Hcy-Lys isopeptide which was discovered in human plasma and can be formed by proteolytic degradation of
N-homocysteinylated proteins (
Figure 4). However, the function of isopeptide and its metabolism is still not known.
N-homocysteinylated proteins exhibit biological responses, including cytotoxicity, immune activation (anti-
N-homocysteinylated proteins IgG antibodies), and atherothrombosis
[31][48]. Anti-
N-homocysteinylated protein autoantibodies can be beneficial by clearing damaged proteins from circulation. However, the autoantibodies can be harmful when they form the antigen–autoantibody complex with
N-homocysteinylated protein on the vascular endothelium. The macrophages will bind to the complex and digest it, causing damage to the vascular wall and tissue damage. If this process were to occur in brain vasculature, it would result in cognitive domain-specific outcomes depending on which brain region was damaged
[48]. Thus, anti-
N-homocysteinylated protein autoantibodies can impair functional, but not structural, aspects of cognition
[48]. The authors
[48] suggest anti-
N-homocysteinylated protein autoantibodies as a risk factor for cognitive impairment.
Several lines of evidence have shown that HTL and
N-homocysteinylated proteins could be causal factors for many various disorders and diseases. Human plasma contains 0.1–13 μM
N-homocysteinylated protein, which represents up to 25% of total plasma Hcy
[34]. Protein
N-homocysteinylation increases significantly in CBS- and MTHFR-deficient patients as up to 30-fold
[30]. Folic acid and vitamin B deficiency diet increases the level of
N-homocysteinylated proteins
[30][40]. Plasma concentrations of
N-homocysteinylated proteins are correlated with total Hcy which allows various factors to influence it. It is not surprising, that protein
N-homocysteinylation is partly reversibly modifiable by a diet
[30]. For example, plasma
N-homocysteinylated protein levels increased 11.6-fold in mice fed a high-methionine diet for 2 weeks, compared with animals fed a normal diet
[30]. After two weeks, the increase in total Hcy, HTL, and
N-homocysteinylated protein were 36-, 14-, and 12-fold, respectively. Replacing the diet to normal for two weeks, the level total Hcy and HTL returned almost to the control/started concentrations. However, the concentration of
N-homocysteinylated proteins remained twice the control
[30]. According to these results, the total Hcy and HTL levels in healthy patients can be decreased quite quickly. To normalize the level of
N-homocysteinylated proteins, much more time is required. Over this time, some irreversible changes may occur. The long-lasting proteins may contain considerably higher degrees of
N-homocysteinylation which may result in unexpected change. These results can explain the failure of Hcy-lowering clinical trials using vitamin B supplementation on total Hcy level in cardiovascular disease. However, the exact mechanism of the clinical trials’ inconsistent results by folic acid and vitamin B supplementation requires additional extended research.
Homocysteinylation represents an emerging class of protein modifications that extremely affect protein function. However, it is still not known the mechanisms involved in N-homocysteinylated protein contribution to the cellular response. Hcy and HTL metabolism is an insufficiently explored area that alters our understanding of the etiology and pathogenesis of the disease. However, the clinical Hcy test does not identify HTL and N-homocysteinylated protein levels, making it difficult for clinical data analysis.
 + credit
+ credit