 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Tomas Hrncir | + 2359 word(s) | 2359 | 2021-05-10 10:46:08 | | | |
| 2 | Camila Xu | -75 word(s) | 2284 | 2021-05-31 03:33:50 | | |
Video Upload Options
The pathogenesis of NAFLD is complex and not fully understood, but there is increasing evidence that the gut microbiota is strongly implicated in the development of NAFLD.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is characterised by an excessive intrahepatic fat accumulation, i.e., steatosis, without significant alcohol consumption. Liver steatosis is defined as fat accumulation, in >5% of hepatocytes [1]. NAFLD may be present in several forms ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), which is a progressive form, characterised by steatosis, hepatocytes swelling, and inflammation. Unlike simple steatosis, NASH is not reversible and can eventually progress into fibrosis, cirrhosis, or even hepatocellular carcinoma (HCC).
The first stage of alcoholic liver disease (ALD) is also characterised by hepatic steatosis. However, unlike NAFLD, the primary trigger of ALD, i.e., excessive alcohol consumption, is known and the disease is preventable. Ethanol probably does not play a prominent role in NAFLD pathogenesis but is discussed as one of the possible contributing factors. A detailed discussion of the role of the gut microbiota in ALD pathogenesis is beyond the scope of this review and has been discussed elsewhere [2].
NAFLD is closely associated with many features of metabolic syndrome, including obesity, insulin resistance, hyperlipidaemia, and hypertension [3][4] and increases the risk of cardiovascular disease (CVD) and type 2 diabetes mellitus (T2DM) [5]. Therefore, not surprisingly, the leading cause of death in NAFLD patients is not liver failure, but cardiovascular disease [6].
NAFLD is the most common chronic liver condition in the USA and Europe. Its global prevalence is rapidly increasing and is currently estimated at 24%. The highest rates are reported from the Middle East (32%) and South America (31%) and the lowest from Africa (14%) [7]. The estimated 10-year economic burden of NAFLD alone could increase to an estimated USD 1.005 trillion in the USA and EUR 334 billion in Europe [8].
NAFLD pathogenesis is complex and not fully understood. The current understanding is that NAFLD is caused by a complex interplay of environmental factors mostly dietary, gut microbiota disturbances, and host factors.
2. Gut Microbiota Dysbiosis
2.1. Introduction to Dysbiosis
Liver diseases, including non-alcoholic fatty liver disease (NAFLD), alcoholic liver disease (ALD), cirrhosis, and hepatocellular carcinoma [9] are associated with compositional and functional alterations of gut microbiota, known as dysbiosis. Dysbiosis is usually characterised by these two major features: (1) the reduction or complete loss of some commensals. The total loss of certain microbial species leads to decreased microbiota diversity, which is associated with many immune-mediated and metabolic disorders [10]. (2) The overgrowth of potentially pathogenic commensals (pathobionts). In a healthy gut ecosystem, pathobionts represent a relatively low percentage of gut microbiota. However, in many diseases, the pathobionts outgrow other commensals. For example, the outgrowth of Gram-negative bacteria from the Enterobacteriaceae family, a subgroup of Proteobacteria phylum, is frequently observed in many immune-mediated and metabolic diseases including NAFLD [11][12]. The bloom of Proteobacteria is often considered as a potential diagnostic marker of dysbiosis and risk of disease [13].
Diverse gut microbiota of each individual may endow the host with unique metabolic apparatus and the ability to adapt to changing environment and substrate availability. With decreasing microbial diversity during urbanisation/industrialisation this adaptability was partially lost as human gut microbiota gained new abilities aimed at sugar and xenobiotics processing [14][15].
2.2. Triggers and Drivers of Dysbiosis
Dysbiosis might be caused by host-derived factors such as genetic background, health status (infection, inflammation), and lifestyle habits or, even more importantly, by environmental factors such as diet (high in sugars, low in fibre), xenobiotics (antibiotics, medication, food additives, chlorinated water), or hygienic environment.
Profound shifts in gut bacterial and fungal microbiota can be quickly achieved with shifts in macronutrients. These changes have significant physiological consequences, as diets rich in animal protein or simple sugars worsen the intestinal inflammation induced by dextran sulphate sodium. However, while the former increases proinflammatory tuning in gut monocytes the latter worsens the gut barrier function. In both cases, however, interactions between diet and microbiota are necessary for this deleterious effects as they fail to appear in germ-free condition or after transfer of the microbiota to naive mice [16][17].
The effect of food additives on gut microbiota has been long overlooked, but recently, several groups, including ours published data demonstrating that some human gut microbes are highly susceptible to food preservatives [18] and that the exposure to common preservatives promotes Proteobacteria overgrowth [19]. Chassaing et al. found that dietary emulsifiers directly alter human gut microbiota composition, and that the emulsifier-modified microbiota can induce intestinal inflammation when transplanted to germ-free mice [20]. Additionally, Rodriguez-Palacios et al. [21] showed that the artificial sweetener Splenda promotes Proteobacteria dysbiosis and increases myeloperoxidase reactivity in ileitis-prone SAMP mice. In addition, stevioside, another noncaloric sweetener, has been shown to cause structural and functional changes in the gut microbiota [22]. Non-caloric artificial sweeteners have been widely used as sugar substitutes, and their main purpose was to decrease energy intake and prevent the development of obesity and metabolic syndrome. Unfortunately, they often induce dysbiosis and drive glucose intolerance in a microbiota-dependent manner, thus inducing deleterious metabolic effects they were aimed to prevent [23].
Host-derived factors modifying gut microbiota load and composition are bactericidal fluids produced by gastric glands and the liver, i.e., gastric acid and bile, and antimicrobial molecules, such as defensins, lysozymes, and antibacterial lectins (Reg3γ) produced by Paneth cells or SIgA produced by plasma cells [24].
2.3. Consequences of Dysbiosis
Dysbiotic microbiota can influence the host immune and metabolic systems and mucosal integrity via various mechanisms. Immune system-modulating mechanisms include the modulation of inflammasome signalling through microbial metabolites, the modulation of Toll-like receptor (TLR) and NOD-like receptor (NLR) signalling, the degradation of secretory IgA (SIgA), the shifting of the balance between regulatory and proinflammatory T cell subsets, direct mucolytic activity and others [25]. Metabolic system-modulating mechanisms include various effects on glucose and lipid metabolism mediated by changes in bile acid composition, the production of short-chain fatty acids (SCFAs) from dietary fibre, the conversion of choline to TMA, and many others [26]. The integrity of the intestinal wall could be disrupted by acetaldehyde produced by microbiota from exogeneous or endogenous ethanol [27], by direct mucolytic activity [28] and other mechanisms.
Whether dysbiosis is a direct cause of NAFLD or merely reflects disease-associated changes in the host’s immune and metabolic systems remains unclear. However, there is accumulating evidence from both preclinical and clinical studies suggesting that gut microbiota dysbiosis plays a key role in the initiation of the disease and its maintenance.
3. NAFLD-Associated Microbiota Signatures
3.1. Gut Microbiota Signatures
The gut microbiota alterations associated with NAFLD are dependent on the clinical stages of the disease [29]. The most typical general characteristics of the disease’s progression include decreasing microbiota diversity, an increasing abundance of Gram-negative bacteria, mostly Proteobacteria (phylum), and a decreasing abundance of Gram-positive bacteria, mostly Firmicutes (phylum) [30]. Functionally, there is also a shift from beneficial to harmful microbes leading to the development of a proinflammatory and metabolically toxic intestinal environment resulting in gut barrier dysfunction, the exposure of the liver to dietary and microbiota-derived factors, and NAFLD progression [29].
The most consistent gut microbiota signatures associated with NAFLD are increased Proteobacteria (phylum), Enterobacteriaceae (family), Escherichia, Bacteroides, Dorea, and Peptoniphilus (genus) and decreased Rikenellaceae, Ruminococcaceae (family), Faecalibacterium, Coprococcus, Anaerosporobacter, and Eubacterium (genera) (Table 1). The NAFLD-associated microbiota signatures partially overlap with other metabolic diseases. For example, the levels of Faecalibacterium prausnitzii are reduced in cirrhosis [31] as well as in obesity [32], T2DM [33], or in intestinal disorders, such as IBD [34] or IBS [35]. F. prausnitzii is considered a beneficial microbe for its anti-inflammatory properties [32]. Another strain, Bacteroides vulgatus, which is increased in advanced fibrosis [30], is associated with severe obesity, insulin resistance, and increased haemoglobin A1c levels [36]. Interestingly, cirrhosis patients often have a higher abundance of oral cavity microbial strains, such as Prevotella, Veillonella, and Streptococcus [31], in the gut microbiome, which are generally not present in healthy individuals.
Unlike patients with ALD who have fungal dysbiosis characterised by decreased diversity and Candida overgrowth [37], patients with NAFLD have no alterations in gut mycobiome.
Table 1. NAFLD-associated gut microbiota signatures.
| Phylum | Class | Family | Genus |
|---|---|---|---|
| Proteobacteria↑ [11][12][30][38][39] | Gammaproteobacteria↑ [40] | Enterobacteriaceae↑ [11][12] | Shigella↑ [11] |
| Escherichia↑ [12][30][38] | |||
| Pasteurellaceae↑ [39] | Haemophilus↓ [38] | ||
| Succinivibrionaceae↑ [41] | |||
| Epsilonproteobacteria↑ [40] | |||
| Alphaproteobacteria | Kiloniellaceae↑ [39] | ||
| Bradyrhizobiaceae | Bradyrhizobium↑ [42] | ||
| Verrucomicrobia↑ [38] | Verrucomicrobiae | Akkermansiaceae | Akkermansia↑ [38] |
| Fusobacteria↑ [11] | |||
| Bacteroidetes↑↓ [11][12][40][42][43][44] | Bacteroidia↑ [43] | Rikenellaceae↓ [12][42] | Alistipes↓ [12] |
| Bacteroidaceae | Bacteroides↑ [45] | ||
| Bacteroidetes | Prevotellaceae↑↓ [11][12] | Prevotella↑↓ [11][12][40][45] | |
| Porphyromonadaceae↑↓ [39][43] | Porphyromonas↑ [12] | ||
| Parabacteroides↑ [41] | |||
| Coprobacter↓ [38] | |||
| Firmicutes↑↓ [12][30][38][39][40][41][42][43] | Clostridia↓ [43] | Streptococcaceae↑ [11] | |
| Clostridiaceae↓ [43] | Anaerotruncus↓ [43] | ||
| Ruminococcaceae↓ [11][12][39][43] | Ruminococcus↑↓ [42][43][45][46][47] | ||
| Flavonifractor↑ [38] | |||
| Subdoligranulum↓ [38] | |||
| Faecalibacterium↓ [12][46][48] | |||
| Oscillospira↓ [42] | |||
| Peptostreptococcaceae↓ [43] | |||
| Lachnospiraceae↑↓ [11][12][38][39][42][43] | Lachnospiraceae incertae sedis↑ [11] | ||
| Robinsoniella↑ [39] | |||
| Dorea↑ [39][42] | |||
| Coprococcus↓ [12][38][43][46][47] | |||
| Moryella↓ [43] | |||
| Pseudobutyrivibrio↓ [43] | |||
| Anaerosporobacter↓ [43] | |||
| Roseburia↑↓ [12][39][43] | |||
| Blautia↑↓ [11][12][42][45] | |||
| Peptoniphilaceae | Peptoniphilus↑ [42][43] | ||
| Clostridiales family XI. incertae sedis | Anaerococcus↑ [42] | ||
| Eubacteriaceae | Eubacterium↓ [12][38] | ||
| Oscillospiraceae | Oscillibacter↑↓ [38][39][42] | ||
| Negativicutes | Veillonellaceae↑ [39] | Allisonella↑ [41] | |
| Erysipelotrichia | Erysipelotrichaceae↑ [11] | Holdemania↓ [38] | |
| Bacilli | Lactobacillaceae↑↓ [43][46] | Lactobacillus↑↓ [39][43][46] | |
| Acidaminococcaceae | Acidaminococcus↑ [38] | ||
| Actinobacteria↑↓ [12][38][42] | Actinobacteria | Bifidobacteriaceae↓ [12] | Bifidobacterium↑↓ [12][38] |
| Coriobacteriaceae | Eggerthella↑ [38] |
↑ = increased, ↓ = decreased, ↑↓ = contradictory, unassigned = data not available.
3.2. Liver and Circulatory Microbiome
Evidence of viable liver or circulating microbiota in NAFLD patients is limited. Nevertheless, bacteria of genera Staphylococcus and Acinetobacter can be cultured from venous blood of cirrhosis patients [49]. These cirrhosis patients also underwent an implantation of intrahepatic portosystemic shunt (TIPS). During the procedure, portal, hepatic, central, and peripheral venous blood samples were collected and the 16S rRNA analysis showed that the most abundant phylum in all four compartments is Proteobacteria (about 90%) [49].
4. Gut–Liver Axis—Bidirectional Link
4.1. Definition
The gut–liver axis is a bidirectional communication through the biliary tract, portal vein, and systemic circulation. Liver-derived factors, such as bile acids, influence gut microbiota composition and function, and gut-derived products, either dietary or microbial, regulate bile acid synthesis and as well as glucose and lipid metabolism in the liver. The disruption of the gut–liver axis by, for example, environmental factors inducing gut dysbiosis and/or increased intestinal permeability leads to proinflammatory changes in the liver, and its failure to regulate gut microbiota results in further disease progression (Figure 1). A comprehensive understanding of gut–liver communication is a key to developing efficient preventative, diagnostic, and therapeutic approaches.
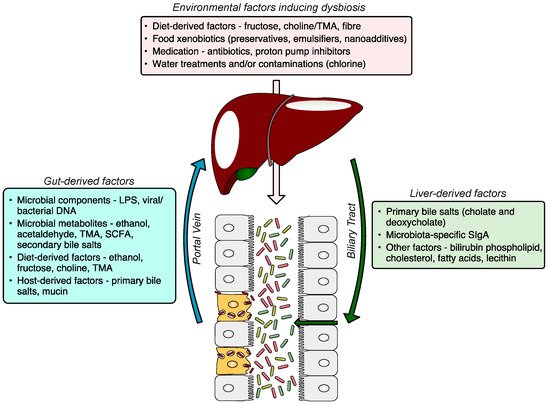
Figure 1. The gut–liver axis and the role of the gut microbiome in the pathogenesis of NAFLD. The gut microbiome altered by environmental factors induces hepatic steatosis. The dysfunctional liver fails to restore gut eubiosis, which results in a vicious self-perpetuating loop, promoting NAFLD progression. The communication via systemic mediators is not shown. NAFLD, non-alcoholic fatty liver disease; MAMP, microbe-associated molecular pattern; LPS, lipopolysaccharide; TMA, trimethylamine; SCFA, short-chain fatty acid; DCA, deoxycholic acid; CDCA, chenodeoxycholic acid.
4.2. Intestinal Barrier Dysfunction
The key function of the barrier is to protect tissues and organs from harmful luminal contents, such as parasites, microorganisms, MAMPs, microbial metabolites, dietary antigens, or toxins while preserving nutrient absorption. The intestinal barrier consists of several functional elements. The physical barrier consists of commensal bacteria, mucins secreted by goblet cells, and the intestinal epithelium sealed with tight junction proteins. The immunological barrier includes components of cellular and humoral immunity. Humoral factors, such as antimicrobial peptides and SIgA, control the load and composition of the microbiota in the lumen. The major antimicrobial peptides are defensins, cathelicidins, resistin-like molecules, and lectins produced by specialised epithelial cells and Paneth cells. SIgA is secreted by plasma cells of the lamina propria, activated by antigen-presenting cells, and transported into the intestinal lumen by epithelial cells after binding to the polymeric Ig receptor (pIgR) [50]. SIgA binds to microbial antigens and toxins, protecting the intestinal mucosa from damage, a process known as immune exclusion [51]. The other immunological components that protect the integrity of the mucosa include macrophages and dendritic cells that transport the infiltrated bacteria and antigens to the mesenteric lymph nodes (MLN), allowing the priming and maturation of B and T cells that form the adaptive immune response in the gut-associated lymphoid tissue [52].
There is no consensus as to which factors are major contributors to increased intestinal permeability, however, there is accumulating evidence that environmental factors, especially an unhealthy diet characterised by low fibre, high sugar and HFCS content, and some food additives play a significant role. For example, chronic fructose consumption is associated with tight junction disruption [53] and increased intestinal permeability [54][55]. The excess unabsorbed fructose is metabolised by gut microbiota which results in lactic acidosis. Other factors compromising the intestinal barrier include excessive alcohol consumption, high exposure to medications including antibiotics, stress, and a lack of physical activity. These factors affect gut permeability either directly or via an induction of gut microbiota dysbiosis.
4.3. Liver and Immune System
The liver is evolutionarily programmed to tolerate low-level exposure to innocuous dietary and microbial antigens delivered via the portal vein. Liver tolerance is maintained by hepatic antigen-presenting cells (HAPCs), which include dendritic cells, liver sinusoidal endothelial cells (LSECs), Kupffer cells, and hepatic stellate cells [56][57]. Antigen presentation by HAPCs to T cells results in suppression of T cell responses [58]. HAPCs also secrete anti-inflammatory cytokines, such as transforming growth factor-beta (TGF-β) and interleukin 10 (IL-10), in response to low levels of microbiota-derived antigens in portal blood. Both TGF-β and IL-10 promote the differentiation of regulatory T cells (Tregs), which suppress the proliferation and effector functions of CD4+ cells and CD8+ T cells [59][60]. These two immunological mechanisms lead to the induction of liver tolerance and have been shown to protect the liver from immune-mediated liver injury [61][62]. However, when the intestinal barrier is compromised, the liver becomes overloaded with antigens from the gut, leading to a loss of liver tolerance and the development of a proinflammatory milieu. Antigens derived from the microbiota induce inflammation by binding to pattern recognition receptors (PRRs) on liver macrophages, including Kupffer cells and stellate cells [63][64]. Signalling via PRRs, mostly TLRs, leads to increased production of inflammatory (TNFα, IL-1, IL-6) and fibrogenic cytokines/chemokines (TGFβ, MCP-1) as well as oxidative and endoplasmic reticulum (ER) stress [65]. Microbial antigens can also induce type I interferon responses in the liver, leading to proliferation and activation of CD8+ cytotoxic T cells [66]. Other immunological mechanisms, such as various effects of short-chain fatty acids on adaptive immune responses, are discussed separately. All of these immunological mechanisms may contribute to the development of inflammation-mediated liver injury, which may progress to fibrosis, cirrhosis, or even HCC.
References
- European Association for the Study of the Liver (EASL); European Association for the Study of Diabetes (EASD); European Association for the Study of Obesity (EASO). EASL-EASD-EASO Clinical Practice Guidelines for the Management of Non-Alcoholic Fatty Liver Disease. Obes. Facts 2016, 9, 65–90.
- Bajaj, J.S. Alcohol, liver disease and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 235–246.
- Ballestri, S.; Nascimbeni, F.; Romagnoli, D.; Lonardo, A. The independent predictors of non-alcoholic steatohepatitis and its individual histological features.: Insulin resistance, serum uric acid, metabolic syndrome, alanine aminotransferase and serum total cholesterol are a clue to pathogenesis and candidate targets for treatment. Hepatol. Res. 2016, 46, 1074–1087.
- Stepanova, M.; Rafiq, N.; Makhlouf, H.; Agrawal, R.; Kaur, I.; Younoszai, Z.; McCullough, A.; Goodman, Z.; Younossi, Z.M. Predictors of All-Cause Mortality and Liver-Related Mortality in Patients with Non-Alcoholic Fatty Liver Disease (NAFLD). Dig. Dis. Sci. 2013, 58, 3017–3023.
- Vanni, E.; Bugianesi, E.; Kotronen, A.; De Minicis, S.; Yki-Järvinen, H.; Svegliati-Baroni, G. From the metabolic syndrome to NAFLD or vice versa? Dig. Liver Dis. 2010, 42, 320–330.
- Athyros, V.G.; Alexandrides, T.K.; Karagiannis, A.; Karvounis, C.; Katsiki, N.; Kotsis, V.; Kountouras, J.; Liberopoulos, E.; Pitsavos, C.; Polyzos, S.; et al. The use of statins alone, or in combination with pioglitazone and other drugs, for the treatment of non-alcoholic fatty liver disease/non-alcoholic steatohepatitis and related cardiovascular risk. An Expert Panel Statement. Metabolism 2017, 71, 17–32.
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84.
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global burden of NAFLD and NASH: Trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20.
- Betrapally, N.S.; Gillevet, P.M.; Bajaj, J.S. Gut microbiome and liver disease. Transl. Res. 2017, 179, 49–59.
- Carding, S.; Verbeke, K.; Vipond, D.T.; Corfe, B.M.; Owen, L.J. Dysbiosis of the gut microbiota in disease. Microb. Ecol. Health Dis. 2015, 26, 26191.
- Shen, F.; Zheng, R.-D.; Sun, X.-Q.; Ding, W.-J.; Wang, X.-Y.; Fan, J.-G. Gut microbiota dysbiosis in patients with non-alcoholic fatty liver disease. Hepatobiliary Pancreat. Dis. Int. 2017, 16, 375–381.
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609.
- Shin, N.-R.; Whon, T.W.; Bae, J.-W. Proteobacteria: Microbial signature of dysbiosis in gut microbiota. Trends Biotechnol. 2015, 33, 496–503.
- Gomez, A.; Petrzelkova, K.J.; Burns, M.B.; Yeoman, C.J.; Amato, K.R.; Vlckova, K.; Modry, D.; Todd, A.; Robinson, C.A.J.; Remis, M.J.; et al. Gut Microbiome of Coexisting BaAka Pygmies and Bantu Reflects Gradients of Traditional Subsistence Patterns. Cell Rep. 2016, 14, 2142–2153.
- Mancabelli, L.; Milani, C.; Lugli, G.A.; Turroni, F.; Ferrario, C.; Van Sinderen, D.; Ventura, M. Meta-analysis of the human gut microbiome from urbanized and pre-agricultural populations. Environ. Microbiol. 2017, 19, 1379–1390.
- Kostovcikova, K.; Coufal, S.; Galanova, N.; Fajstova, A.; Hudcovic, T.; Kostovcik, M.; Prochazkova, P.; Zakostelska, Z.J.; Cermakova, M.; Sediva, B.; et al. Diet Rich in Animal Protein Promotes Pro-inflammatory Macrophage Response and Exacerbates Colitis in Mice. Front. Immunol. 2019, 10, 919.
- Fajstova, A.; Galanova, N.; Coufal, S.; Malkova, J.; Kostovcik, M.; Cermakova, M.; Pelantova, H.; Kuzma, M.; Sediva, B.; Hudcovic, T.; et al. Diet Rich in Simple Sugars Promotes Pro-Inflammatory Response via Gut Microbiota Alteration and TLR4 Signaling. Cells 2020, 9, 2701.
- Hrncirova, L.; Machova, V.; Trckova, E.; Krejsek, J.; Hrncir, T. Food Preservatives Induce Proteobacteria Dysbiosis in Human-Microbiota Associated Nod2-Deficient Mice. Microorganisms 2019, 7, 383.
- Hrncirova, L.; Hudcovic, T.; Sukova, E.; Machova, V.; Trckova, E.; Krejsek, J.; Hrncir, T. Human gut microbes are susceptible to antimicrobial food additives in vitro. Folia Microbiol. 2019, 64, 497–508.
- Chassaing, B.; Van De Wiele, T.; De Bodt, J.; Marzorati, M.; Gewirtz, A.T. Dietary emulsifiers directly alter human microbiota composition and gene expression ex vivo potentiating intestinal inflammation. Gut 2017, 66, 1414–1427.
- Rodriguez-Palacios, A.; Harding, A.; Menghini, P.; Himmelman, C.; Retuerto, M.; Nickerson, K.P.; Lam, M.; Croniger, C.M.; McLean, M.H.; Durum, S.K.; et al. The Artificial Sweetener Splenda Promotes Gut Proteobacteria, Dysbiosis, and Myeloperoxidase Reactivity in Crohn’s Disease–Like Ileitis. Inflamm. Bowel Dis. 2018, 24, 1005–1020.
- Gatea, F.; Sârbu, I.; Vamanu, E. In Vitro Modulatory Effect of Stevioside, as a Partial Sugar Replacer in Sweeteners, on Human Child Microbiota. Microorganisms 2021, 9, 590.
- Suez, J.; Korem, T.; Kuperman, Y.; Harmelin, A.; Kolodkin-Gal, I.; Shapiro, H.; Halpern, Z.; Segal, E.; Elinav, E.; Zeevi, D.; et al. Artificial sweeteners induce glucose intolerance by altering the gut microbiota. Nature 2014, 514, 181–186.
- Catanzaro, J.R.; Strauss, J.D.; Bielecka, A.; Porto, A.F.; Lobo, F.M.; Urban, A.; Schofield, W.B.; Palm, N.W. IgA-deficient humans exhibit gut microbiota dysbiosis despite secretion of compensatory IgM. Sci. Rep. 2019, 9, 1–10.
- Levy, M.; Kolodziejczyk, A.A.; Thaiss, C.A.; Elinav, E. Dysbiosis and the immune system. Nat. Rev. Immunol. 2017, 17, 219–232.
- Chu, H.; Duan, Y.; Yang, L.; Schnabl, B. Small metabolites, possible big changes: A microbiota-centered view of non-alcoholic fatty liver disease. Gut 2018, 68, 359–370.
- Llorente, C.; Schnabl, B. The Gut Microbiota and Liver Disease. Cell. Mol. Gastroenterol. Hepatol. 2015, 1, 275–284.
- Png, C.W.; Lindén, S.K.; Gilshenan, K.S.; Zoetendal, E.G.; McSweeney, C.S.; Sly, L.I.; McGuckin, M.A.; Florin, T.H.J. Mucolytic Bacteria with Increased Prevalence in IBD Mucosa Augment In Vitro Utilization of Mucin by Other Bacteria. Am. J. Gastroenterol. 2010, 105, 2420–2428.
- Aron-Wisnewsky, J.; Vigliotti, C.; Witjes, J.; Le, P.; Holleboom, A.G.; Verheij, J.; Nieuwdorp, M.; Clément, K. Gut microbiota and human NAFLD: Disentangling microbial signatures from metabolic disorders. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 279–297.
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5.
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64.
- Le Chatelier, E.; Nielsen, T.; Qin, J.; Prifti, E.; Hildebrand, F.; Falony, G.; Almeida, M.; Arumugam, M.; Batto, J.-M.; Kennedy, S.; et al. Richness of human gut microbiome correlates with metabolic markers. Nature 2013, 500, 541–546.
- Karlsson, F.H.; Tremaroli, V.; Nookaew, I.; Bergström, G.; Behre, C.J.; Fagerberg, B.; Nielsen, J.; Bäckhed, F. Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 2013, 498, 99–103.
- Sokol, H.; Seksik, P.; Furet, J.P.; Firmesse, O.; Nion-Larmurier, I.; Beaugerie, L.; Cosnes, J.; Corthier, G.; Marteau, P.; Doré, J. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm. Bowel Dis. 2009, 15, 1183–1189.
- Rajilić–Stojanović, M.; Biagi, E.; Heilig, H.G.; Kajander, K.; Kekkonen, R.A.; Tims, S.; de Vos, W.M. Global and Deep Molecular Analysis of Microbiota Signatures in Fecal Samples From Patients with Irritable Bowel Syndrome. Gastroenterology 2011, 141, 1792–1801.
- Aron-Wisnewsky, J.; Prifti, E.; Belda, E.; Ichou, F.; Kayser, B.D.; Dao, M.C.; Verger, E.O.; Hedjazil, L.; Bouillot, J.-L.; Chevallier, J.-M.; et al. Major microbiota dysbiosis in severe obesity: Fate after bariatric surgery. Gut 2019, 68, 70–82.
- Yang, A.-M.; Inamine, T.; Hochrath, K.; Chen, P.; Wang, L.; Llorente, C.; Bluemel, S.; Hartmann, P.; Xu, J.; Koyama, Y.; et al. Intestinal fungi contribute to development of alcoholic liver disease. J. Clin. Investig. 2017, 127, 2829–2841.
- Hoyles, L.; Fernandez-Real, J.-M.; Federici, M.; Serino, M.; Abbott, J.; Charpentier, J.; Heymes, C.; Luque, J.L.; Anthony, E.; Barton, R.H. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat. Med. 2018, 24, 1070–1080.
- Raman, M.; Ahmed, I.; Gillevet, P.M.; Probert, C.S.; Ratcliffe, N.M.; Smith, S.; Greenwood, R.; Sikaroodi, M.; Lam, V.; Crotty, P.; et al. Fecal Microbiome and Volatile Organic Compound Metabolome in Obese Humans with Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2013, 11, 868–875.e3.
- Michail, S.; Lin, M.; Frey, M.R.; Fanter, R.; Paliy, O.; Hilbush, B.; Reo, N.V. Altered gut microbial energy and metabolism in children with non-alcoholic fatty liver disease. FEMS Microbiol. Ecol. 2014, 91, 1–9.
- Wong, V.W.-S.; Tse, C.-H.; Lam, T.T.-Y.; Wong, G.L.-H.; Chim, A.M.-L.; Chu, W.C.-W.; Yeung, D.K.-W.; Law, P.T.-W.; Kwan, H.-S.; Yu, J.; et al. Molecular Characterization of the Fecal Microbiota in Patients with Nonalcoholic Steatohepatitis–A Longitudinal Study. PLoS ONE 2013, 8, e62885.
- Del Chierico, F.; Nobili, V.; Vernocchi, P.; Russo, A.; De Stefanis, C.; Gnani, D.; Furlanello, C.; Zandonà, A.; Paci, P.; Capuani, G.; et al. Gut microbiota profiling of pediatric nonalcoholic fatty liver disease and obese patients unveiled by an integrated meta-omics-based approach. Hepatology 2017, 65, 451–464.
- Wang, B.; Jiang, X.; Cao, M.; Ge, J.; Bao, Q.; Tang, L.; Chen, Y.; Li, L. Altered Fecal Microbiota Correlates with Liver Biochemistry in Nonobese Patients with Non-alcoholic Fatty Liver Disease. Sci. Rep. 2016, 6, 32002.
- Mouzaki, M.; Comelli, E.M.; Arendt, B.M.; Bonengel, J.; Fung, S.K.; Fischer, S.E.; McGilvray, I.D.; Allard, J.P. Intestinal microbiota in patients with nonalcoholic fatty liver disease. Hepatology 2013, 58, 120–127.
- Boursier, J.; Mueller, O.; Hunault, G.; Oberti, F.; Calès, P.; Diehl, A.M.; Barret, M.; Machado, M.V.; Fizanne, L.; Araujo-Perez, F.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775.
- Da Silva, H.E.; Teterina, A.; Comelli, E.M.; Taibi, A.; Arendt, B.M.; Fischer, S.E.; Lou, W.; Allard, J.P. Nonalcoholic fatty liver disease is associated with dysbiosis independent of body mass index and insulin resistance. Sci. Rep. 2018, 8, 1–12.
- Alferink, L.J.; Radjabzadeh, D.; Erler, N.S.; Vojinovic, D.; Medina-Gomez, C.; Uitterlinden, A.G.; de Knegt, R.J.; Amin, N.; Ikram, M.A.; Janssen, H.L.; et al. Microbiomics, Metabolomics, Predicted Metagenomics, and Hepatic Steatosis in a Population-Based Study of 1355 Adults. Hepatology 2021, 73, 968–982.
- Chen, Y.; Yang, F.; Lu, H.; Wang, B.; Chen, Y.; Lei, D.; Wang, Y.; Zhu, B.; Li, L. Characterization of fecal microbial communities in patients with liver cirrhosis. Hepatology 2011, 54, 562–572.
- Schierwagen, R.; Alvarez-Silva, C.; Madsen, M.S.A.; Kolbe, C.C.; Meyer, C.; Thomas, D.; Uschner, F.E.; Magdaleno, F.; Jansen, C.; Pohlmann, A.; et al. Circulating microbiome in blood of different circulatory compartments. Gut 2019, 68, 578–580.
- Brandtzaeg, P.; Prydz, H. Direct evidence for an integrated function of J chain and secretory component in epithelial transport of immunoglobulins. Nature 1984, 311, 71–73.
- Mestecky, J.; Russell, M.W.; Elson, C.O. Intestinal IgA: Novel views on its function in the defence of the largest mucosal surface. Gut 1999, 44, 2–5.
- Gautreaux, M.D.; Deitch, E.A.; Berg, R.D. T lymphocytes in host defense against bacterial translocation from the gastrointestinal tract. Infect. Immun. 1994, 62, 2874–2884.
- Johnson, R.J.; Rivard, C.; Lanaspa, M.A.; Otabachian-Smith, S.; Ishimoto, T.; Cicerchi, C.; Cheeke, P.R.; MacIntosh, B.; Hess, T. Fructokinase, Fructans, Intestinal Permeability, and Metabolic Syndrome: An Equine Connection? J. Equine Veter. Sci. 2013, 33, 120–126.
- Spruss, A.; Bergheim, I. Dietary fructose and intestinal barrier: Potential risk factor in the pathogenesis of nonalcoholic fatty liver disease. J. Nutr. Biochem. 2009, 20, 657–662.
- Bergheim, I.; Weber, S.; Vos, M.; Krämer, S.; Volynets, V.; Kaserouni, S.; McClain, C.J.; Bischoff, S.C. Antibiotics protect against fructose-induced hepatic lipid accumulation in mice: Role of endotoxin. J. Hepatol. 2008, 48, 983–992.
- Crispe, I.N. Liver antigen-presenting cells. J. Hepatol. 2011, 54, 357–365.
- Horst, A.K.; Neumann, K.; Diehl, L.; Tiegs, G. Modulation of liver tolerance by conventional and nonconventional antigen-presenting cells and regulatory immune cells. Cell. Mol. Immunol. 2016, 13, 277–292.
- Karimi, M.H.; Geramizadeh, B.; Malek-Hosseini, S.A. Tolerance Induction in Liver. Int. J. Organ Transplant. Med. 2015, 6, 45–54.
- Breous, E.; Somanathan, S.; Vandenberghe, L.H.; Wilson, J.M. Hepatic regulatory T cells and Kupffer cells are crucial mediators of systemic T cell tolerance to antigens targeting murine liver. Hepatology 2009, 50, 612–621.
- Carambia, A.; Freund, B.; Schwinge, D.; Heine, M.; Laschtowitz, A.; Huber, S.; Wraith, D.C.; Korn, T.; Schramm, C.; Lohse, A.W.; et al. TGF-β-dependent induction of CD4+CD25+Foxp3+ Tregs by liver sinusoidal endothelial cells. J. Hepatol. 2014, 61, 594–599.
- Crispe, I.N. Immune tolerance in liver disease. Hepatology 2014, 60, 2109–2117.
- Doherty, D.G. Antigen-specific immune tolerance in the liver. Nat. Biomed. Eng. 2019, 3, 763–765.
- Isayama, F.; Hines, I.N.; Kremer, M.; Milton, R.J.; Byrd, C.L.; Perry, A.W.; McKim, S.E.; Parsons, C.; Rippe, R.A.; Wheeler, M.D. LPS signaling enhances hepatic fibrogenesis caused by experimental cholestasis in mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2006, 290, G1318–G1328.
- Gäbele, E.; Mühlbauer, M.; Dorn, C.; Weiss, T.S.; Froh, M.; Schnabl, B.; Wiest, R.; Schölmerich, J.; Obermeier, F.; Hellerbrand, C. Role of TLR9 in hepatic stellate cells and experimental liver fibrosis. Biochem. Biophys. Res. Commun. 2008, 376, 271–276.
- Lebeaupin, C.; Proics, E.; De Bieville, C.H.D.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879.
- Ghazarian, M.; Revelo, X.S.; Nøhr, M.K.; Luck, H.; Zeng, K.; Lei, H.; Tsai, S.; Schroer, S.A.; Park, Y.J.; Chng, M.H.Y.; et al. Type I interferon responses drive intrahepatic T cells to promote metabolic syndrome. Sci. Immunol. 2017, 2, eaai7616.


