The pathogenesis of NAFLD is complex and not fully understood, but there is increasing evidence that the gut microbiota is strongly implicated in the development of NAFLD.
The pathogenesis of NAFLD is complex and not fully understood, but there is increasing evidence that the gut microbiota is strongly implicated in the development of NAFLD. In this review, we discuss the major factors that induce dysbiosis of the gut microbiota and disrupt intestinal permeability, as well as possible mechanisms leading to the development of NAFLD. We also discuss the most consistent NAFLD-associated gut microbiota signatures and immunological mechanisms involved in maintaining the gut barrier and liver tolerance to gut-derived factors. Finally, we review currently available diagnostic and prognostic methods, summarise latest knowledge on promising microbiota-based biomarkers, and discuss therapeutic strategies
- liver steatosis
- cirrhosis
- hepatocellular carcinoma
- intestinal permeability
- gut microbiota
- dysbiosis
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is characterised by an excessive intrahepatic fat accumulation, i.e., steatosis, without significant alcohol consumption. Liver steatosis is defined as fat accumulation, in >5% of hepatocytes [1]. NAFLD may be present in several forms ranging from simple steatosis to non-alcoholic steatohepatitis (NASH), which is a progressive form, characterised by steatosis, hepatocytes swelling, and inflammation. Unlike simple steatosis, NASH is not reversible and can eventually progress into fibrosis, cirrhosis, or even hepatocellular carcinoma (HCC).
The first stage of alcoholic liver disease (ALD) is also characterised by hepatic steatosis. However, unlike NAFLD, the primary trigger of ALD, i.e., excessive alcohol consumption, is known and the disease is preventable. Ethanol probably does not play a prominent role in NAFLD pathogenesis but is discussed as one of the possible contributing factors. A detailed discussion of the role of the gut microbiota in ALD pathogenesis is beyond the scope of this review and has been discussed elsewhere [2].
NAFLD is closely associated with many features of metabolic syndrome, including obesity, insulin resistance, hyperlipidaemia, and hypertension [3][4] and increases the risk of cardiovascular disease (CVD) and type 2 diabetes mellitus (T2DM) [5]. Therefore, not surprisingly, the leading cause of death in NAFLD patients is not liver failure, but cardiovascular disease [6].
NAFLD is closely associated with many features of metabolic syndrome, including obesity, insulin resistance, hyperlipidaemia, and hypertension [3,4] and increases the risk of cardiovascular disease (CVD) and type 2 diabetes mellitus (T2DM) [5]. Therefore, not surprisingly, the leading cause of death in NAFLD patients is not liver failure, but cardiovascular disease [6].
NAFLD is the most common chronic liver condition in the USA and Europe. Its global prevalence is rapidly increasing and is currently estimated at 24%. The highest rates are reported from the Middle East (32%) and South America (31%) and the lowest from Africa (14%) [7]. The estimated 10-year economic burden of NAFLD alone could increase to an estimated USD 1.005 trillion in the USA and EUR 334 billion in Europe [8].
NAFLD pathogenesis is complex and not fully understood. The current understanding is that NAFLD is caused by a complex interplay of environmental factors mostly dietary, gut microbiota disturbances, and host factors.
2. Gut Microbiota Dysbiosis
2.1. Introduction to Dysbiosis
Enterobacteriaceae family, a subgroup of Proteobacteria phylum, is frequently observed in many immune-mediated and metabolic diseases including NAFLD [11][12]. The bloom of Proteobacteria is often considered as a potential diagnostic marker of dysbiosis and risk of disease [13].
Diverse gut microbiota of each individual may endow the host with unique metabolic apparatus and the ability to adapt to changing environment and substrate availability. With decreasing microbial diversity during urbanisation/industrialisation this adaptability was partially lost as human gut microbiota gained new abilities aimed at sugar and xenobiotics processing [14][15].
2.2. Triggers and Drivers of Dysbiosis
Profound shifts in gut bacterial and fungal microbiota can be quickly achieved with shifts in macronutrients. These changes have significant physiological consequences, as diets rich in animal protein or simple sugars worsen the intestinal inflammation induced by dextran sulphate sodium. However, while the former increases proinflammatory tuning in gut monocytes the latter worsens the gut barrier function. In both cases, however, interactions between diet and microbiota are necessary for this deleterious effects as they fail to appear in germ-free condition or after transfer of the microbiota to naive mice [16][17].
2.3. Consequences of Dysbiosis
3. NAFLD-Associated Microbiota Signatures
3.1. Gut Microbiota Signatures
Enterobacteriaceae
Escherichia
Bacteroides
Dorea
Peptoniphilus
Rikenellaceae
Ruminococcaceae
Faecalibacterium
Coprococcus
Anaerosporobacter
Eubacterium
Faecalibacterium prausnitzii
F. prausnitzii
Bacteroides vulgatus
Prevotella
Veillonella
Streptococcus
Candida
Table 1.
| Phylum | Class | Family | Genus | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Proteobacteria↑ [11][12][30][38][39] | Proteobacteria↑ [11,12,30,38,39] | Gammaproteobacteria↑ [40] | Enterobacteriaceae↑ [11][12] | ↑ [11,12] | Shigella | ↑ [11] | |||||||
| Escherichia↑ [12][30][38] | ↑ [12,30,38] | ||||||||||||
| Pasteurellaceae | ↑ [39] | Haemophilus | ↓ [38] | ||||||||||
| Succinivibrionaceae | ↑ [41] | ||||||||||||
| Epsilonproteobacteria↑ [40] | |||||||||||||
| Alphaproteobacteria | Kiloniellaceae | ↑ [39] | |||||||||||
| Bradyrhizobiaceae | Bradyrhizobium | ↑ [42] | |||||||||||
| Verrucomicrobia↑ [38] | Verrucomicrobiae | Akkermansiaceae | Akkermansia | ↑ [38] | |||||||||
| Fusobacteria↑ [11] | |||||||||||||
| Bacteroidetes↑↓ [11][12][40][42][43][44] | Bacteroidetes↑↓ [11,12,40,42,43,44] | Bacteroidia↑ [43] | Rikenellaceae↓ [12][42] | ↓ [12,42] | Alistipes | ↓ [12] | |||||||
| Bacteroidaceae | Bacteroides | ↑ [45] | |||||||||||
| Bacteroidetes | Prevotellaceae↑↓ [11][12] | ↑↓ [11,12] | Prevotella↑↓ [11][12][40][45] | ↑↓ [11,12,40,45] | |||||||||
| Porphyromonadaceae↑↓ [39][43] | ↑↓ [39,43] | Porphyromonas | ↑ [12] | ||||||||||
| Parabacteroides | ↑ [41] | ||||||||||||
| Coprobacter | ↓ [38] | ||||||||||||
| Firmicutes↑↓ [12][30][38][39][40[43] | Firmicutes↑↓ [12,30 | ][ | ,38 | 41][ | ,39 | 42 | ,40 | ] | ,41,42,43] | Clostridia↓ [43] | Streptococcaceae | ↑ [11] | |
| Clostridiaceae | ↓ [43] | Anaerotruncus | ↓ [43] | ||||||||||
| Ruminococcaceae↓ [11][12][39] | ↓ [11 | [ | ,12 | 43 | ,39 | ] | ,43] | Ruminococcus↑↓ [42][43][][46][47] | ↑↓ [42,43 | 45 | ,45,46,47] | ||
| Flavonifractor | ↑ [38] | ||||||||||||
| Subdoligranulum | ↓ [38] | ||||||||||||
| Faecalibacterium↓ [12][46][48] | ↓ [12,46,48] | ||||||||||||
| Oscillospira | ↓ [42] | ||||||||||||
| Peptostreptococcaceae | ↓ [43] | ||||||||||||
| Lachnospiraceae↑↓ [11][12][38][39][42][43] | ↑↓ [11,12,38,39,42,43] | Lachnospiraceae incertae sedis↑ [11] | |||||||||||
| Robinsoniella | ↑ [39] | ||||||||||||
| Dorea↑ [39][42] | ↑ [39,42] | ||||||||||||
| Coprococcus↓ [12][38][43][46][47] | ↓ [12,38,43,46,47] | ||||||||||||
| Moryella | ↓ [43] | ||||||||||||
| Pseudobutyrivibrio | ↓ [43] | ||||||||||||
| Anaerosporobacter | ↓ [43] | ||||||||||||
| Roseburia↑↓ [12][39][43] | ↑↓ [12,39,43] | ||||||||||||
| Blautia↑↓ [11][12][42][45] | ↑↓ [11,12,42,45] | ||||||||||||
| Peptoniphilaceae | Peptoniphilus↑ [42][43] | ↑ [42,43] | |||||||||||
| Clostridiales family XI. incertae sedis | Anaerococcus | ↑ [42] | |||||||||||
| Eubacteriaceae | Eubacterium↓ [12][38] | ↓ [12,38] | |||||||||||
| Oscillospiraceae | Oscillibacter↑↓ [38][39][42] | ↑↓ [38,39,42] | |||||||||||
| Negativicutes | Veillonellaceae | ↑ [39] | Allisonella | ↑ [41] | |||||||||
| Erysipelotrichia | Erysipelotrichaceae | ↑ [11] | Holdemania | ↓ [38] | |||||||||
| Bacilli | Lactobacillaceae↑↓ [43] | ↑↓ [43 | [46] | ,46] | Lactobacillus↑↓ [39][43][46] | ↑↓ [39,43,46] | |||||||
| Acidaminococcaceae | Acidaminococcus | ↑ [38] | |||||||||||
| Actinobacteria↑↓ [12][38][42] | Actinobacteria↑↓ [12,38,42] | Actinobacteria | Bifidobacteriaceae | ↓ [12] | Bifidobacterium↑↓ [12][38] | ↑↓ [12,38] | |||||||
| Coriobacteriaceae | Eggerthella | ↑ [38] |
↑ = increased, ↓ = decreased, ↑↓ = contradictory, unassigned = data not available.
3.2. Liver and Circulatory Microbiome
3.2. Liver and Circulatory Microbiome
Staphylococcus
Acinetobacter
4. Gut–Liver Axis—Bidirectional Link
4.1. Definition
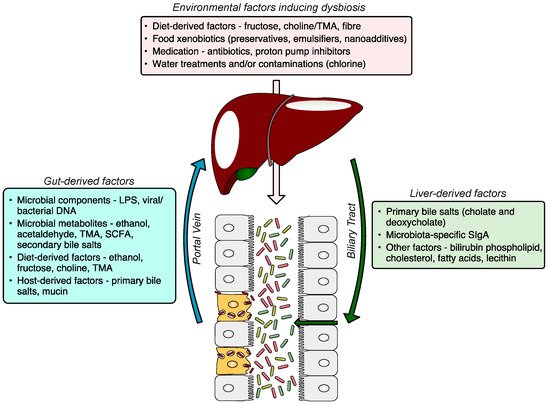
Figure 1.
4.2. Intestinal Barrier Dysfunction
There is no consensus as to which factors are major contributors to increased intestinal permeability, however, there is accumulating evidence that environmental factors, especially an unhealthy diet characterised by low fibre, high sugar and HFCS content, and some food additives play a significant role. For example, chronic fructose consumption is associated with tight junction disruption [53] and increased intestinal permeability [54][55]. The excess unabsorbed fructose is metabolised by gut microbiota which results in lactic acidosis. Other factors compromising the intestinal barrier include excessive alcohol consumption, high exposure to medications including antibiotics, stress, and a lack of physical activity. These factors affect gut permeability either directly or via an induction of gut microbiota dysbiosis.
4.3. Liver and Immune System
The liver is evolutionarily programmed to tolerate low-level exposure to innocuous dietary and microbial antigens delivered via the portal vein. Liver tolerance is maintained by hepatic antigen-presenting cells (HAPCs), which include dendritic cells, liver sinusoidal endothelial cells (LSECs), Kupffer cells, and hepatic stellate cells [56][57]. Antigen presentation by HAPCs to T cells results in suppression of T cell responses [58]. HAPCs also secrete anti-inflammatory cytokines, such as transforming growth factor-beta (TGF-β) and interleukin 10 (IL-10), in response to low levels of microbiota-derived antigens in portal blood. Both TGF-β and IL-10 promote the differentiation of regulatory T cells (Tregs), which suppress the proliferation and effector functions of CD4+ cells and CD8+ T cells [59][60]. These two immunological mechanisms lead to the induction of liver tolerance and have been shown to protect the liver from immune-mediated liver injury [61][62]. However, when the intestinal barrier is compromised, the liver becomes overloaded with antigens from the gut, leading to a loss of liver tolerance and the development of a proinflammatory milieu. Antigens derived from the microbiota induce inflammation by binding to pattern recognition receptors (PRRs) on liver macrophages, including Kupffer cells and stellate cells [63][64]. Signalling via PRRs, mostly TLRs, leads to increased production of inflammatory (TNFα, IL-1, IL-6) and fibrogenic cytokines/chemokines (TGFβ, MCP-1) as well as oxidative and endoplasmic reticulum (ER) stress [65]. Microbial antigens can also induce type I interferon responses in the liver, leading to proliferation and activation of CD8+ cytotoxic T cells [66]. Other immunological mechanisms, such as various effects of short-chain fatty acids on adaptive immune responses, are discussed separately. All of these immunological mechanisms may contribute to the development of inflammation-mediated liver injury, which may progress to fibrosis, cirrhosis, or even HCC.
The liver is evolutionarily programmed to tolerate low-level exposure to innocuous dietary and microbial antigens delivered via the portal vein. Liver tolerance is maintained by hepatic antigen-presenting cells (HAPCs), which include dendritic cells, liver sinusoidal endothelial cells (LSECs), Kupffer cells, and hepatic stellate cells [56,57]. Antigen presentation by HAPCs to T cells results in suppression of T cell responses [58]. HAPCs also secrete anti-inflammatory cytokines, such as transforming growth factor-beta (TGF-β) and interleukin 10 (IL-10), in response to low levels of microbiota-derived antigens in portal blood. Both TGF-β and IL-10 promote the differentiation of regulatory T cells (Tregs), which suppress the proliferation and effector functions of CD4+ cells and CD8+ T cells [59,60]. These two immunological mechanisms lead to the induction of liver tolerance and have been shown to protect the liver from immune-mediated liver injury [61,62]. However, when the intestinal barrier is compromised, the liver becomes overloaded with antigens from the gut, leading to a loss of liver tolerance and the development of a proinflammatory milieu. Antigens derived from the microbiota induce inflammation by binding to pattern recognition receptors (PRRs) on liver macrophages, including Kupffer cells and stellate cells [63,64]. Signalling via PRRs, mostly TLRs, leads to increased production of inflammatory (TNFα, IL-1, IL-6) and fibrogenic cytokines/chemokines (TGFβ, MCP-1) as well as oxidative and endoplasmic reticulum (ER) stress [65]. Microbial antigens can also induce type I interferon responses in the liver, leading to proliferation and activation of CD8+ cytotoxic T cells [66]. Other immunological mechanisms, such as various effects of short-chain fatty acids on adaptive immune responses, are discussed separately. All of these immunological mechanisms may contribute to the development of inflammation-mediated liver injury, which may progress to fibrosis, cirrhosis, or even HCC.