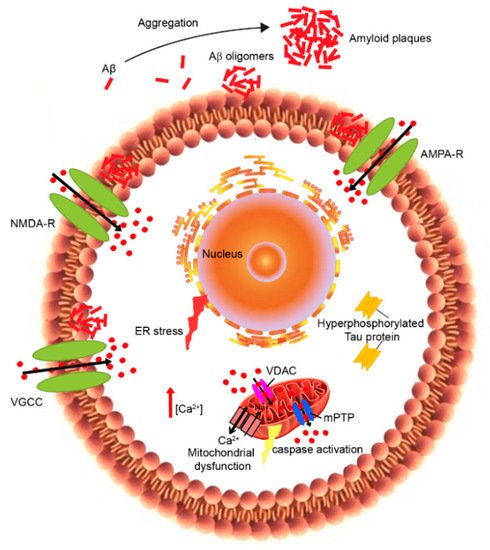
The so-called “Calcium hypothesis” was first postulated by Khachaturian in 1989
[1] following important experimental studies by the group of
[2][3]. It explored how the activation of the amyloidogenic pathway may remodel the neuronal Ca
2+ signaling pathways responsible for cognition. According to this hypothesis, the depolarization of aged neurons causes the influx of Ca
2+ from the extracellular space and excitotoxicity. Other studies instead demonstrated that neuronal aging is associated with the alteration of neuronal Ca
2+ extrusion, leading to old neurons being more vulnerable
[4][5][6]. In particular, Ca
2+ dyshomeostasis has been reported in both peripheral and central neurons during the aging process
[4][6] as well as in neurons of AD patients
[7][8], influencing both Aβ production and Tau hyperphosphorylation
[9][10] (1).
Indeed, several studies performed on both AD brains and experimental models showed that the alteration of Ca
2+ homeostasis occurs before the development of the symptoms, suggesting that Ca
2+ dysregulation is an upstream event in AD pathogenesis
[11]. In addition, the calcium overload was coupled to the deposition of senile plaques and was most pronounced in the immediate vicinity of senile plaques in transgenic mouse models
[12][13]. In particular, increased cytosolic Ca
2+ levels can promote Aβ production and its following neurotoxicity, while the accumulation of the Aβ peptide results in the stimulation of neuronal Ca
2+ signaling
[14]. Therefore, a synergic mechanism between Ca
2+ and Aβ could intensify the neurodegeneration and cognitive deficits in AD patients
[15].
2. Plasma Membrane Calcium Dysregulation
The ability of aberrant protein oligomers to penetrate and disrupt the cellular membrane and induce toxicity appears to result from direct interactions with the lipid bilayers
[16][17][18][19][20]. Trodusquemine, a natural product in the aminosterol class, was recently shown to enhance the rate of Aβ aggregation, thus reducing the lifetime or number of toxic oligomeric species. In addition, trodusquemine functions at phisiological concentration to prevent Aβ toxicity by displacyng the aggregates from the cell membranes
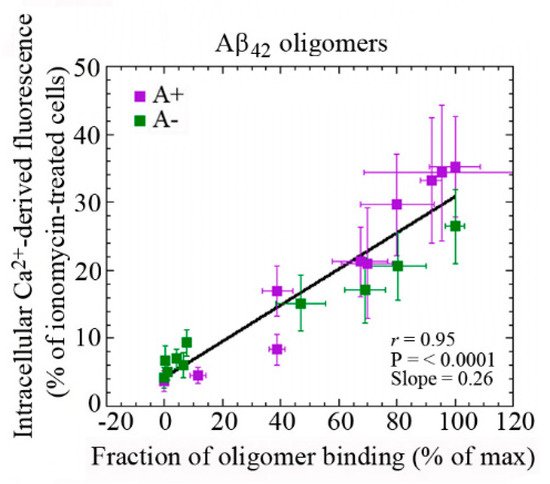
[19][20]. These studies provide confidence that aminosterols could be useful in the treatment of AD. Our previous analysis has shown the existence of a linear correlation between the rate of Ca
2+ influx across plasma membranes and the amount of oligomeric species bound to the neuronal surface ()
[21]. These findings indicate that the susceptibility of neuronal cells to different types of misfolded oligomers is directly related to the extent of the binding of such oligomers to the cellular membrane. The ability of cell membranes to bind oligomeric aggregates appears to depend in turn on the physicochemical properties of both the oligomers and the membranes, which for the latter are determined in large part by their lipid composition
[18][22][23]. In particular, the monosialotetrahexosylganglioside GM1 has been found to be an important factor in the context of AD
[21][24][25]. GM1, together with cholesterol and sphingomyelin, is abundant in lipid raft domains within the cell membrane that contain a vast array of membrane proteins, including channels and receptors
[26].
Figure 2. Dependence of intracellular Ca
2+ dyshomeostasis on the binding affinities of Aβ42 oligomers to cellular membranes with different GM1 content. Changes in the intracellular Ca
2+ levels plotted against the fraction of oligomer binding to the plasma membrane in GM1-modulated SH-SY5Y neuroblastoma cells treated for 1 h with 10 μM A+ (violet) or A− (green) oligomers of Aβ42 formed according to Ladiwala’s protocol
[23]. Reprinted from 2B, Evangelisti et al., 2016, licensed under Creative Commons Attribution 4.0 International Public License (CC BY 4.0;
https://creativecommons.org/licenses/by/4.0/ (accessed on 5 May 2021))
[21].
There is strong evidence of a key role for PrPc, a protein that is associated with lipid rafts, as a receptor for oligomers of the Aβ peptide, resulting in the activation of a Fyn-mediated complex signaling cascade leading to tau phosphorylation and Ca
2+ dyshomeostasis
[27]. Several studies support the idea that oligomers can interact with membranes through direct binding to GM1
[28][29]. This then results in the disruption of lipid bilayers, the alteration of their permeability and the malfunction of raft-associated Ca
2+ channels, leading to Ca
2+ influx into cells.
Although there is a variety of experiential evidence, the amyloid channel hypothesis still remains controversial. Indeed, numerous mechanisms can be responsible for the Aβ interaction with neuronal membranes causing the disruption of Ca
2+ homeostasis. These include the activation of some type of cell surface receptor coupled to Ca
2+ influx, and the alteration of membrane permeability
[18][30]. Several reports showed that alterations in Ca
2+ levels cause the dysfunction of VGCCs
[6], the downregulation of Ca
2+ clearance mechanisms at the plasma membrane level
[4][6] and the failure of Ca
2+ homeostatic machinery located on intracellular organelles
[5][6]. These events affected the maintenance of Ca
2+ signals in old neurons, impairing learning and memory. It has been shown that Aβ can stimulate the opening of VGCCs, which, in turn, increases the intracellular concentration of Ca
2+ [31]. In addition, the increase in intracellular Ca
2+ levels can stimulate the overexpression of Ltype calcium channel subtype (Cav 1.2) in the hippocampal cell membranes of AD models, causing the influx of Ca
2+ [32]. The inhibition of SERCA as well as the release of Ca
2+ via the RyR caused the increase in cytoplasmic Ca
2+. This overloading caused the activation of β-secretase and thus an increased Aβ production and aggregation
[33].
In the past few years, in vitro studies demonstrated that the Aβ peptide formed cation-selective pores into the plasma membrane, thus causing Ca
2+ influx from the extracellular space across these Aβ pore-channels
[34][35][36][37]. However, other in vivo studies showed that Aβ can improve the plasma membrane permeability to both anions and cations by altering its dielectric structure
[38]. Following this evidence, numerous researchers have focused their attention on the effects of Aβ peptides on Ca
2+ channels in neuronal cells. In particular, blocking Ca
2+ influx was found to reduce the neurotoxicity of Aβ oligomers and the levels of insoluble Aβ1–40 and Aβ1–42 in the hippocampus of AD transgenic mice
[39]. The increase in intracellular Ca
2+ promotes the activation of CaN and that of the phosphatases, including PP1, which is involved in long-term depression (LTD)
[40]. CaN can contribute together with Aβ or tau to the loss of dendritic spines and synapses, leading to cognitive deficit in AD mouse models. Accordingly, CaN inhibitors can reverse or improve these impairments
[41][42]. The Ca
2+/CaM complex activates the CaMKII, playing an important role in memory formation and synaptic plasticity. Taking into account that many kinases can be activated by Ca
2+, the dyshomeostasis of Ca
2+ can increase tau phosphorylation
[43][44][45]. Conversely, abnormal accumulation of intracellular tau can also induce Ca
2+ overload, causing the dephosphorylation of CaMKIV and CREB by activating CaN
[41]. The increase in cytosolic Ca
2+ can also activate JAK2-STAT1 signaling, leading to the binding of STAT1 to NMDARs, and thus inhibits the transcription of specific GAS elements
[43]; the increase in the cleaved STAT1 induced by tau also activates BACE1, promoting Aβ production
[46]. All of these alterations reveal new mechanisms by which tau can induce synapse impairments and cognitive deficits. PS1 and PS2 have also been implicated in the influx of Ca
2+, as well as in the ER and mitochondrial Ca
2+ signaling. Indeed, mutations in these proteins affect Ca
2+ homeostasis
[47].
An altered expression of calcium-binding (CBP) and calcium sensing proteins can modify the calcium-buffering capacity, causing the oversensitivity of neurons to glutamate released in the extracellular space
[48][49][50]. In addition, the impairment of glutamate transporter function in the glial cell leads to the accumulation of glutamate in the synaptic cleft, activating the postsynaptic AMPA/NMDA receptors. Thus, Ca
2+ entry in postsynaptic neurons disrupts the intracellular homeostasis and induces an increase in ROS production. Aβ has been reported to bind to NMDA and AMPA glutamate receptors
[51], as well as nicotinic acetylcholine receptors
[52], and all of these receptors are highly Ca
2+-permeable. Furthermore, Aβ can influence VGCCs and IP
3R
[12]. Numerous research studies demonstrated that Aβ promotes the upregulation of VGCCs in different neuronal types, including cortical and hippocampal neurons
[53]. In particular, Aβ peptides may over-activate the function of L-type VGCCs through a mechanism involving ROS production
[54], or induce the overexpression of CaV1.2 and CaV1.3 channels in the hippocampus of AD transgenic mice
[55] and in rat hippocampal neurons treated with Aβ
[56]. The effects of Aβ on NMDARs have attracted considerable interest as these ligand-gated channels are involved in synaptic plasticity and LTP
[57]. Indeed, Aβ oligomers can induce the overactivation of NMDARs, resulting in a cytosolic Ca
2+ increase
[21][5][25][58][59] through several mechanisms: by affecting glutamate availability
[60][61] and/or by modifying NMDAR electrophysiological properties
[62], or by changing membrane tension
[63]. Importantly, the overactivation of GluN2B NMDAR subunits induced by Aβ
[64] has been correlated to ER stress, to the depolarization and dysfunction of mitochondria
[65][66], to microtubule disassembly and to a reduction in neurite length
[64]. The synaptic activation of NMDAR is crucial for memory formation
[67]. Hippocampal neurons showed differential expression of NMDA receptor subunits (NR1, NR2B) in AD-like rats
[68]. In particular, the NR2B subunit, which is highly selective for Ca
2+ transport and is known to play a decisive role in Ca
2+-induced apoptosis, was overexpressed in AD models compared to controls
[68]. The persistent overactivation of the NMDA receptor in the postsynaptic terminal stimulates hippocampal neurons, which allows higher calcium influx resulting in excitotoxicity. The Aβ-induced activation of NMDAR promoted its endocytosis
[69]. We have recently observed that lysophosphatidylcholine and arachidonic acid, which cause membrane compression and stretch, respectively, can activate NMDAR and AMPAR through a change in membrane tension induced by Aβ oligomers
[63]. In particular, lysophosphatidylcholine is able to neutralize the oligomer-induced activation of the NMDA receptors, whereas arachidonic acid activates the receptors similarly to the oligomers with no additive effects, suggesting that Aβ-induced toxicity can also be caused by the perturbation of the mechanical properties of lipid membranes sensed by NMDA and AMPA receptors
[63]. Memantine, an NMDAR antagonist, was approved in 2002 as a therapeutic drug in moderate to severe AD
[70][71]. However, other potential drugs targeting NMDARs are not included in AD therapy because of their intolerable side effects.
4. Mitochondrial Calcium Dyshomeostasis
Mitochondria play a key role in the modulation of intracellular Ca
2+ signaling
[87]. Indeed, they can quickly resume Ca
2+ to prevent Ca
2+ overload into the cytosol, activating the Ca
2+-dependent mitochondrial matrix dehydrogenase to produce ATP. However, this disruption of mitochondrial Ca
2+ regulation affects energy production and oxidative stress, resulting in mitochondrial dysfunction
[88]. In particular, the excessive Ca
2+ influx into the mitochondria induces mitochondrial outer membrane permeabilization and the subsequent release of pro-apoptotic factors into the cytoplasm, including cytochrome C and apoptosis-inducing factor, which activate apoptosis cell death
[89] (). Mitochondrial dysfunction has been proposed as an early event in AD and other aging-related neurodegenerative disorders
[90]. Studies on brains from AD patients and AD mouse models showed impaired mitochondrial function, associated with decreased bioenergetics and ATP synthesis
[91], morphological abnormalities
[92], the imbalance of mitochondrial dynamics
[93] and the redistribution of mitochondria
[94]. Synapses are particularly rich in mitochondria, which provide energy for Ca
2+ homeostasis. In addition, synaptic mitochondria are more sensitive to Ca
2+ dyshomeostasis with respect to non-synaptic mitochondria
[95]. During LTP, microtubule associated protein 1B (MAP1B) phosphorylation and local concentrations of CaMKII were increased
[96]. CaMKII is responsible for phosphorylating MAP2, which enhances synaptic response
[97]. The accumulation of tau impairs synapses and memory by activating Ca
2+-dependent CaN and suppressing nuclear CaMKIV/CREB signaling, thus revealing a new mechanism by which tau can induce synaptic toxicity
[41]. In vitro studies have reported that Aβ oligomers induce mitochondrial Ca
2+ uptake
[98][99] and Ca
2+ transfer from the ER to mitochondria
[100] in cultured rat primary neurons, even if the in vivo mechanism remains unknown. A recent study showed that the increase in Ca
2+ levels in neuronal mitochondria of transgenic mice appeared only after plaque deposition and before neural death
[101]. This mitochondrial Ca
2+ overload involves toxic extracellular Aβ oligomers and requires the mitochondrial Ca
2+ uniporter. The authors propose a novel potential therapeutic target for AD through the blocking of the mitochondrial Ca
2+ uniporter that was found to reduce mitochondrial Ca
2+ overload
[101].
Various studies have observed that the increase in Ca
2+ levels in the ER caused the leakage of Ca
2+ into the cytoplasmic compartment, affecting the mitochondrial calcium homeostasis
[8][102]. Recently, an interesting hypothesis has been proposed about the role of MAM, the lipid raft-like domain of the ER closely opposed to mitochondria (), in AD pathogenesis
[103]. As already mentioned, MAM is involved in several important mechanisms, including calcium transport, the synthesis of phospholipids, mitochondrial fission and fusion, the division of mtDNA, and cholesterol esterification
[104]. Thus, several studies have been investigating the role of mitochondrial dysfunction mediated by Aβ and MAM, underlying its putative role in AD.
 +1 point
+1 point