Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder that is characterized by amyloid β-protein deposition in senile plaques, neurofibrillary tangles consisting of abnormally phosphorylated tau protein, and neuronal loss leading to cognitive decline and dementia. Despite extensive research, the exact mechanisms underlying AD remain unknown and effective treatment is not available. Many hypotheses have been proposed to explain AD pathophysiology; however, there is general consensus that the abnormal aggregation of the amyloid β peptide (Aβ) is the initial event triggering a pathogenic cascade of degenerating events in cholinergic neurons. The dysregulation of calcium homeostasis has been studied considerably to clarify the mechanisms of neurodegeneration induced by Aβ. Intracellular calcium acts as a second messenger and plays a key role in the regulation of neuronal functions, such as neural growth and differentiation, action potential, and synaptic plasticity. The calcium hypothesis of AD posits that activation of the amyloidogenic pathway affects neuronal Ca2+ homeostasis and the mechanisms responsible for learning and memory. Aβ can disrupt Ca2+ signaling through several mechanisms, by increasing the influx of Ca2+ from the extracellular space and by activating its release from intracellular stores. Here, we review the different molecular mechanisms and receptors involved in calcium dysregulation in AD and possible therapeutic strategies for improving the treatment.
- protein aggregation
- amyloid β peptide (Aβ)
- toxic oligomers
- amyloid fibrils
- tau protein
- neurodegeneration
- ionic dysregulation
- glutamatergic receptors
- NMDA
- AMPA
The so-called “Calcium hypothesis” was first postulated by Khachaturian in 1989 [1] following important experimental studies by the group of Landfield [2,3].
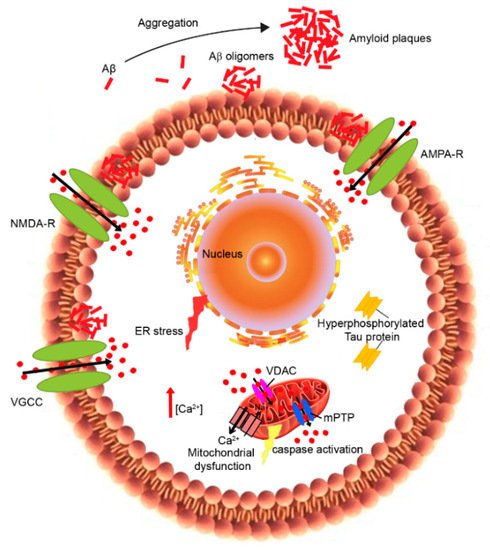
It explored how the activation of the amyloidogenic pathway may remodel the neuronal Ca2+ signaling pathways responsible for cognition. According to this hypothesis, the depolarization of aged neurons causes the influx of Ca2+ from the extracellular space and excitotoxicity. Other studies instead demonstrated that neuronal aging is associated with the alteration of neuronal Ca2+ extrusion, leading to old neurons being more vulnerable [4-6]. In particular, Ca2+ dyshomeostasis has been reported in both peripheral and central neurons during the aging process [4,6] as well as in neurons of Alzheimer’s disease (AD) patients [7,8], influencing both amyloid-β (Aβ) production and Tau hyperphosphorylation [9,10] (Figure 1).
Indeed, several studies performed on both AD brains and experimental models showed that the alteration of calcium ions (Ca2+) homeostasis occurs before the development of the symptoms, suggesting that Ca2+ dysregulation is an upstream event in AD pathogenesis [11]. In addition, the calcium overload was coupled to the deposition of senile plaques and was most pronounced in the immediate vicinity of senile plaques in transgenic mouse models [12,13]. In particular, increased cytosolic Ca2+ levels can promote Aβ production and its following neurotoxicity, while the accumulation of the Aβ peptide results in the stimulation of neuronal Ca2+ signaling [14]. Therefore, a synergic mechanism between Ca2+ and Aβ could intensify the neurodegeneration and cognitive deficits in AD patients [15].
Plasma Membrane Calcium Dysregulation
The ability of aberrant protein oligomers to penetrate and disrupt the cellular membrane and induce toxicity appears to result from direct interactions with the lipid bilayers [16-20]. Trodusquemine, a natural product in the aminosterol class, was recently shown to enhance the rate of Aβ aggregation, thus reducing the lifetime or number of toxic oligomeric species. In addition, trodusquemine functions at phisiological concentration to prevent Aβ toxicity by displacyng the aggregates from the cell membranes [19,20]. These studies provide confidence that aminosterols could be useful in the treatment of AD. Our previous analysis has shown the existence of a linear correlation between the rate of Ca2+ influx across plasma membranes and the amount of oligomeric species bound to the neuronal surface (Figure 2) [21]. These findings indicate that the susceptibility of neuronal cells to different types of misfolded oligomers is directly related to the extent of the binding of such oligomers to the cellular membrane. The ability of cell membranes to bind oligomeric aggregates appears to depend in turn on the physicochemical properties of both the oligomers and the membranes, which for the latter are determined in large part by their lipid composition [18, 22,23]. In particular, the monosialotetrahexosylganglioside 1 (GM1) has been found to be an important factor in the context of AD [21,24,25]. GM1, together with cholesterol and sphingomyelin, is abundant in lipid raft domains within the cell membrane that contain a vast array of membrane proteins, including channels and receptors [26].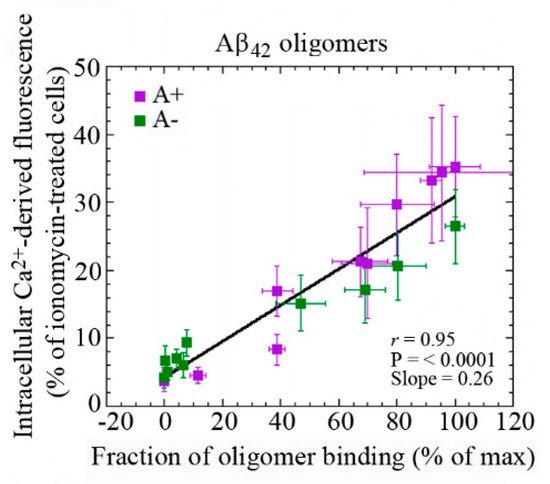
There is strong evidence of a key role for cellular prion protein (PrPc), a protein that is associated with lipid rafts, as a receptor for oligomers of the Aβ peptide, resulting in the activation of a tyrosine-protein kinase (Fyn)-mediated complex signaling cascade leading to tau phosphorylation and Ca2+ dyshomeostasis [27]. Several studies support the idea that oligomers can interact with membranes through direct binding to GM1 [28,29]. This then results in the disruption of lipid bilayers, the alteration of their permeability and the malfunction of raft-associated Ca2+ channels, leading to Ca2+ influx into cells.
Although there is a variety of experiential evidence, the amyloid channel hypothesis still remains controversial. Indeed, numerous mechanisms can be responsible for the Aβ interaction with neuronal membranes causing the disruption of Ca2+ homeostasis. These include the activation of some type of cell surface receptor coupled to Ca2+ influx, and the alteration of membrane permeability [18,30]. Several reports showed that alterations in Ca2+ levels cause the dysfunction of voltage-gated calcium channels (VGCCs) [6], the downregulation of Ca2+ clearance mechanisms at the plasma membrane level [3,6] and the failure of Ca2+ homeostatic machinery located on intracellular organelles [5,6]. These events affected the maintenance of Ca2+ signals in old neurons, impairing learning and memory. It has been shown that Aβ can stimulate the opening of VGCCs, which, in turn, increases the intracellular concentration of Ca2+ [31]. In addition, the increase in intracellular Ca2+ levels can stimulate the overexpression of Ltype calcium channel subtype (Cav 1.2) in the hippocampal cell membranes of AD models, causing the influx of Ca2+ [32]. The inhibition of sarco-endoplasmic reticulum calcium transport ATPase (SERCA) as well as the release of Ca2+ via the ryanodine receptor (RyR) caused the increase in cytoplasmic Ca2+. This overloading caused the activation of β-secretase and thus an increased Aβ production and aggregation [33].
In the past few years, in vitro studies demonstrated that the Aβ peptide formed cation-selective pores into the plasma membrane, thus causing Ca2+ influx from the extracellular space across these Aβ pore-channels [34-37]. However, other in vivo studies showed that Aβ can improve the plasma membrane permeability to both anions and cations by altering its dielectric structure [38]. Following this evidence, numerous researchers have focused their attention on the effects of Aβ peptides on Ca2+ channels in neuronal cells. In particular, blocking Ca2+ influx was found to reduce the neurotoxicity of Aβ oligomers and the levels of insoluble Aβ1–40 and Aβ1–42 in the hippocampus of AD transgenic mice [39]. The increase in intracellular Ca2+ promotes the activation of calcineurin (CaN) and that of the phosphatases, including PP1, which is involved in long-term depression (LTD) [40]. CaN can contribute together with Aβ or tau to the loss of dendritic spines and synapses, leading to cognitive deficit in AD mouse models. Accordingly, CaN inhibitors can reverse or improve these impairments [41,42]. The Ca2+/calmodulin(CaM) complex activates the calmodulin dependent protein kinase II (CaMKII), playing an important role in memory formation and synaptic plasticity. Taking into account that many kinases can be activated by Ca2+, the dyshomeostasis of Ca2+ can increase tau phosphorylation [43-45]. Conversely, abnormal accumulation of intracellular tau can also induce Ca2+ overload, causing the dephosphorylation of calmodulin dependent protein kinase IV (CaMKIV) and cAMP response element binding protein (CREB) by activating CaN [41]. The increase in cytosolic Ca2+ can also activate JAK2-STAT1 signaling, leading to the binding of STAT1 to N-methyl D-aspartate receptors (NMDARs), and thus inhibits the transcription of specific GAS elements [43]; the increase in the cleaved STAT1 induced by tau also activates β-site amyloid precursor protein cleaving enzyme 1 (BACE1), promoting Aβ production [46]. All of these alterations reveal new mechanisms by which tau can induce synapse impairments and cognitive deficits. The presenilin proteins (PS1 and PS2) have also been implicated in the influx of Ca2+, as well as in the ER and mitochondrial Ca2+ signaling. Indeed, mutations in these proteins affect Ca2+ homeostasis [47].
An altered expression of calcium-binding (CBP) and calcium sensing proteins can modify the calcium-buffering capacity, causing the oversensitivity of neurons to glutamate released in the extracellular space [48-50]. In addition, the impairment of glutamate transporter function in the glial cell leads to the accumulation of glutamate in the synaptic cleft, activating the postsynaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)/NMDA receptors. Thus, Ca2+ entry in postsynaptic neurons disrupts the intracellular homeostasis and induces an increase in ROS production. Aβ has been reported to bind to NMDA and AMPA glutamate receptors [51], as well as nicotinic acetylcholine receptors [52], and all of these receptors are highly Ca2+-permeable. Furthermore, Aβ can influence VGCCs and the inositol 1,4, 5-trisphosphate receptor (IP3R) [12]. Numerous research studies demonstrated that Aβ promotes the upregulation of VGCCs in different neuronal types, including cortical and hippocampal neurons [53]. In particular, Aβ peptides may over-activate the function of L-type VGCCs through a mechanism involving ROS production [54], or induce the overexpression of calcium channel subtypes (CaV1.2 and CaV1.3) in the hippocampus of AD transgenic mice [55] and in rat hippocampal neurons treated with Aβ [56]. The effects of Aβ on NMDARs have attracted considerable interest as these ligand-gated channels are involved in synaptic plasticity and long-term potentiation (LTP) [57]. Indeed, Aβ oligomers can induce the overactivation of NMDARs, resulting in a cytosolic Ca2+ increase [5, 21,25,58,59] through several mechanisms: by affecting glutamate availability [60,61] and/or by modifying NMDAR electrophysiological properties [62], or by changing membrane tension [63]. Importantly, the overactivation of GluN2B NMDAR subunits induced by Aβ [64] has been correlated to ER stress, to the depolarization and dysfunction of mitochondria [65,66], to microtubule disassembly and to a reduction in neurite length [64]. The synaptic activation of NMDAR is crucial for memory formation [67]. Hippocampal neurons showed differential expression of NMDA receptor subunits (NR1, NR2B) in AD-like rats [68]. In particular, the NR2B subunit, which is highly selective for Ca2+ transport and is known to play a decisive role in Ca2+-induced apoptosis, was overexpressed in AD models compared to controls [68]. The persistent overactivation of the NMDA receptor in the postsynaptic terminal stimulates hippocampal neurons, which allows higher calcium influx resulting in excitotoxicity. The Aβ-induced activation of NMDAR promoted its endocytosis [69]. We have recently observed that lysophosphatidylcholine and arachidonic acid, which cause membrane compression and stretch, respectively, can activate NMDAR and AMPAR through a change in membrane tension induced by Aβ oligomers [63]. In particular, lysophosphatidylcholine is able to neutralize the oligomer-induced activation of the NMDA receptors, whereas arachidonic acid activates the receptors similarly to the oligomers with no additive effects, suggesting that Aβ-induced toxicity can also be caused by the perturbation of the mechanical properties of lipid membranes sensed by NMDA and AMPA receptors [63]. Memantine, an NMDAR antagonist, was approved in 2002 as a therapeutic drug in moderate to severe AD [70,71]. However, other potential drugs targeting NMDARs are not included in AD therapy because of their intolerable side effects.
Endoplasmic-Reticulum and Mitochondrial Calcium Dysregulation
As reported above, Aβ peptides are able to induce a high release of Ca2+ from the ER, thus promoting the activation of the unfolded protein response (UPR) and endoplasmic reticulum (ER) stress [72,73]. Notably, Aβ oligomers may disrupt ER Ca2+ homeostasis indirectly by the increase in Ca2+ influx from the extracellular space, which in turn triggers Ca2+ release from intracellular stores [74], or directly by interacting with several regulators of ER Ca2+, such as RyR and I3PR [75-80]. On the other hand, ER Ca2+ dyshomeostasis causes abnormal Aβ production and neuronal apoptosis [81-83]. In addition, IP3R was found to modulate Ca2+ homeostasis in AD [84] and its alterations have been detected in cells derived from AD patients since 1994 [85,86].
Mitochondria play a key role in the modulation of intracellular Ca2+ signaling [87]. Indeed, they can quickly resume Ca2+ to prevent Ca2+ overload into the cytosol, activating the Ca2+-dependent mitochondrial matrix dehydrogenase to produce ATP. However, this disruption of mitochondrial Ca2+ regulation affects energy production and oxidative stress, resulting in mitochondrial dysfunction [88]. In particular, the excessive Ca2+ influx into the mitochondria induces mitochondrial outer membrane permeabilization and the subsequent release of pro-apoptotic factors into the cytoplasm, including cytochrome C and apoptosis-inducing factor, which activate apoptosis cell death [89] (Figure 1). Mitochondrial dysfunction has been proposed as an early event in AD and other aging-related neurodegenerative disorders [90]. Studies on brains from AD patients and AD mouse models showed impaired mitochondrial function, associated with decreased bioenergetics and ATP synthesis [91], morphological abnormalities [92], the imbalance of mitochondrial dynamics [93] and the redistribution of mitochondria [94]. Synapses are particularly rich in mitochondria, which provide energy for Ca2+ homeostasis. In addition, synaptic mitochondria are more sensitive to Ca2+ dyshomeostasis with respect to non-synaptic mitochondria [95]. During LTP, microtubule associated protein 1B (MAP1B) phosphorylation and local concentrations of CaMKII were increased [96]. CaMKII is responsible for phosphorylating MAP2, which enhances synaptic response [97]. The accumulation of tau impairs synapses and memory by activating Ca2+-dependent CaN and suppressing nuclear CaMKIV/CREB signaling, thus revealing a new mechanism by which tau can induce synaptic toxicity [41]. In vitro studies have reported that Aβ oligomers induce mitochondrial Ca2+ uptake [98,99] and Ca2+ transfer from the ER to mitochondria [100] in cultured rat primary neurons, even if the in vivo mechanism remains unknown. A recent study showed that the increase in Ca2+ levels in neuronal mitochondria of transgenic mice appeared only after plaque deposition and before neural death [101]. This mitochondrial Ca2+ overload involves toxic extracellular Aβ oligomers and requires the mitochondrial Ca2+ uniporter. The authors propose a novel potential therapeutic target for AD through the blocking of the mitochondrial Ca2+ uniporter that was found to reduce mitochondrial Ca2+ overload [101].
Various studies have observed that the increase in Ca2+ levels in the ER caused the leakage of Ca2+ into the cytoplasmic compartment, affecting the mitochondrial calcium homeostasis [8,102]. Recently, an interesting hypothesis has been proposed about the role of mitochondria-associated ER membrane (MAM), the lipid raft-like domain of the ER closely opposed to mitochondria, in AD pathogenesis [103]. As already mentioned, MAM is involved in several important mechanisms, including calcium transport, the synthesis of phospholipids, mitochondrial fission and fusion, the division of mtDNA, and cholesterol esterification [104]. Thus, several studies have been investigating the role of mitochondrial dysfunction mediated by Aβ and MAM, underlying its putative role in AD.
Concluding Remarks
Many studies have revealed that the perturbation of Ca2+ homeostasis is an early event in the cascade of neuronal alterations underlying the cytotoxicity induced by misfolded Aβ aggregates and hyperphosphorylated tau. However, so far, no common consensus has been reached on the molecular mechanisms of neuronal Ca2+ overload, causing the remodeling of signaling pathways with excitotoxicity and memory dysfunction in AD. The effects of Aβ on NMDARs have attracted considerable interest as these ligand-gated channels are involved in synaptic plasticity and LTP. Importantly, the over-activation of GluN2B NMDAR subunits induced by Aβ has been correlated to ER stress and to the depolarization and dysfunction of mitochondria. Recently, ER and mitochondrial Ca2+ dyshomeostasis have also been proposed as early causative events in AD.
The main unsolved issue is whether the neuronal Ca2+ alterations caused by Aβ extracellular deposits could be non-specific, involving just lipid membrane components, or specific by membrane receptors and other cell surface proteins. There is strong evidence of a key role for PrPc, associated with lipid rafts, as a receptor for Aβ oligomers, resulting in the activation of a Fyn-mediated complex signaling cascade, leading to tau phosphorylation and loss of Ca2+ homeostasis. However, the data reported in several other studies support the idea that Aβ oligomers can interact with membranes through direct binding to GM1. This then results in the disruption of lipid bilayers, the alteration of their permeability and the misfunction of raft-associated Ca2+ channels, leading to Ca2+ influx into cells. These findings do not necessarily contradict the view that PrPc behaves as a receptor of a class of Aβ oligomers. Considering the existence of many structurally distinct conformers, different Aβ aggregates could interact with PrPc and GM1 with different affinities. It is increasingly evident that toxicity is not a feature that is inherent to a given type of misfolded protein oligomer, but is instead a property that emerges from the complex interplay between the structural features of oligomers and the lipid composition of the neuronal membranes. Trodusquemine, a natural product in the aminosterol class, was recently shown to prevent Aβ toxicity by displacyng the aggregates from the cell membranes, suggesting that molecules that interact directly with cell membranes, rather than binding oligomeric aggregate themselves, could represent a useful approach in the treatment of AD.
Overall, the question of the molecular basis of Ca2+ perturbation in AD pathophysiology needs further investigations for the development of targeted therapies for AD. Memantine, an NMDAR antagonist approved in 2002 as a therapeutic drug in moderate to severe AD, appears to be promising in AD therapy, together with other potential drugs targeting NMDARs and PrPc. However, according to the latest evidence that both Aβ and tau pathologies have synergistic effects, the most efficacious approach to slow AD may be to combine anti-Aβ and anti-tau therapies.
Author Contributions: Writing, reviewing, and editing, R.C. and C.C.; conceptualization and supervision, R.C. and C.C.; both authors have read and agreed to the published version of the manuscript.
Funding: This research was funded by the University of Florence (Fondi Ateneo) and by the Ministry of Education, Universities and Research of Italy (Progetto Dipartimento di Eccellenza “Gender Medicine”).
Conflicts of Interest: The authors declare no conflict of interest.
References
- Khachaturian, Z.S. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. N. Y. Acad. Sci. 1989, 568, 1–4.
- Landfield, P.W.; Pitler, T.A. Prolonged Ca2+-dependent after hyperpolarizations in hippocampal neurons of aged rats. Science 1984, 226, 1–4.
- Landfield, P.W. Increased calcium current’ hypothesis of brain aging. Aging 1987, 8, 346–347.
- Verkhratsky, A.; Toescu, E.C. Calcium and neuronal ageing. Trends Neurosci. 1998, 21, 2–7.
- Toescu, E.C.; Verkhratsky, A.; Landfield, P.W. Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci. 2004, 27, 614–620.
- Toescu, E.C.; Verkhratsky, A. The importance of being subtle: Small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 2007, 6, 267–273.
- Foster, T.C. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6, 319–325.
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100.
- Querfurth, H.W.; Selkoe, D.J. Calcium ionophore increases amyloid β peptide production by cultured cells. Biochemistry 1994, 33, 4550–4561.
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463.
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.D.; Alkon, L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Dis. 1998, 5, 37–45.
- Latulippe, J.; Lotito, D. A mathematical model for the effects of amyloid beta on intracellular calcium. PLoS ONE 2018, 13, e0202503.
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225.
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94.
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. Biol. Chem. 2005, 280, 17294–17300.
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Chem. 2013, 182, 30–43.
- Zampagni, M.; Evangelisti, E.; Cascella, R.; Liguri, G.; Becatti, M.; Pensalfini, A.; Uberti, D.; Cenini, G.; Memo, M.; Bagnoli, S.; et al. Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. Mol. Med. (Berl). 2010, 88, 597-608.
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. Cell Sci. 2012, 125, 2416–2427.
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Commun. 2019, 10, 225.
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Biol. 2020, 3, 435.
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Rep. 2016, 6, 32721.
- Evangelisti, E.; Wright, D.; Zampagni, M.; Cascella, R.; Fiorillo, C.; Bagnoli, S.; Relini, A.; Nichino, D.; Scartabelli, T.; Nacmias, B.; et al. Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer’s disease. Alzheimer Res. 2013, 10, 143–153.
- Ladiwala, A.R.; Litt, J.; Kane, R.S.; Aucoin, D.S.; Smith, S.O.; Ranjan, S.; Davis, J.; Van Nostrand, W.E.; Tessier, P.M. Conformational differences between two amyloid beta oligomers of similar size and dissimilar toxicity. Biol. Chem. 2012, 287, 24765–24773.
- Malchiodi-Albedi, F.; Contrusciere, V.; Raggi, C.; Fecchi, K.; Rainaldi, G.; Paradisi, S.; Matteucci, A.; Santini, M.T.; Sargiacomo, M.; Frank, C.; et al. Lipid raft disruption protects mature neurons against amyloid oligomer toxicity. Biophys. Acta 2010, 1802, 406–415.
- Cascella, R.; Evangelisti, E.; Bigi, A.; Becatti, M.; Fiorillo, C.; Stefani, M.; Chiti, F.; Cecchi, C. Soluble oligomers require a ganglioside to trigger neuronal calcium overload. Alzheimers Dis. 2017, 60, 923–938.
- Schengrund, C.L. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 2010, 82, 7–17.
- Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Neurosci. 2012, 15, 1227–1235.
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Abeta oligomers are rapidly sequestered from brain ASF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319.
- Calamai, M.; Evangelisti, E.; Cascella, R.; Parenti, N.; Cecchi, C.; Stefani, M.; Pavone, F. Single molecule experiments emphasize GM1 as a key player of the different cytotoxicity of structurally distinct Aβ1-42 oligomers. Biophys. Acta 2016, 1858, 386–392.
- Fonseca, A.C.R.G.; Moreira, P.I.; Oliveira, C.R.; Cardoso, S.M.; Pinton, P.; Pereira, C.F. Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Neurobiol. 2015, 51, 610–622.
- Ranjan, B.; Chong, K.H.; Zheng, J. Composite mathematical modeling of calcium signaling behind neuronal cell death in Alzheimer’s disease. BMC Syst. Biol. 2018, 12, 10.
- Yang, L.; Wang, Z.; Wang, B.; Justice, N.J.; Zheng, H. Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. Neurosci. 2009, 29, 15660–15668.
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. Alzheimers Dis. Parkinsonism 2017, 7, 374.
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. Alzheimers Dis. Parkinsonism 2017, 7, 374.
- Bhatia, R.; Lin, H.; Lal, R. Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: Evidence for Abeta P channel-mediated cellular toxicity. FASEB J. 2000, 14, 1233–1243.
- Kagan, B.L.; Hirakura, Y.; Azimov, R.; Azimova, R.; Lin, M.C. The channel hypothesis of Alzheimer’s disease: Current status. Peptides 2002, 23, 1311–1315.
- Arispe, N.; Diaz, J.C.; Simakova, O. Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biophys. Acta 2007, 1768, 1952–1965.
- Sokolov, Y.; Kozak, J.A.; Kayed, R.; Chanturiya, A.; Glabe, C.; Hall, J.E. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. Gen. Physiol. 2006, 128, 637–647.
- Samad, N.; Ishaq, S.; Bano, S.; Manzoor, N. Calcium Regulation in Alzheimer’s Disease: Mechanistic Understanding. Coll. Physicians Surg. Pak. 2017, 27, 566–571.
- Reese, L.C.; Taglialatela, G. A role for calcineurin in Alzheimer’s disease. Neuropharmacol. 2011, 9, 685–692.
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Natl. Acad. Sci. USA 2016, 113, E3773–E3781.
- Cavallucci, V.; Berretta, N.; Nobili, A.; Nisticò, R.; Mercuri, N.B.; D’Amelio, M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. NeuroMolecular Med. 2013, 15, 541–548.
- Li, X.G.; Hong, X.Y.; Wang, Y.L.; Zhang, S.J.; Zhang, J.F.; Li, X.C.; Liu, Y.C.; Sun, D.S.; Feng, Q.; Ye, J.W.; et al. Tau accumulation triggers STAT1-dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep. 2019, 20, e47202.
- Lin, L.; Cao, J.; Yang, S.S.; Fu, Z.Q.; Zeng, P.; Chu, J.; Ning, L.N.; Zhang, T.; Shi, Y.; Tian, Q.; et al. Endoplasmic reticulum stress induces spatial memory deficits by activating GSK-3. Cell. Mol. Med. 2018, 22, 3489–3502.
- Liu, Z.C.; Chu, J.; Lin, L.; Song, J.; Ning, L.N.; Luo, H.B.; Yang, S.; Shi, Y.; Wang, Q.; Qu, N.; Zhang, Q.; Wang, J.Z.; Tian, Q. SIL1 rescued bip elevation-related tau hyperphosphorylation in ER stress. Neurobiol. 2016, 53, 983–994.
- Zhang, Z.; Li, X.G.; Wang, Z.H.; Song, M.; Yu, S.P.; Kang, S.S.; Liu, X.; Zhang, Z.; Xie, M.; Liu, G.P.; et al. δ-Secretase-cleaved Tau stimulates Aβ production via upregulating STAT1-BACE1 signaling in Alzheimer’s disease. Psychiatry 2021, 26, 586–603.
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular calcium dysregulation by the Alzheimer’s disease-linked protein presenilin 2. J. Mol. Sci. 2020, 21, 770.
- Gaspar, P.; Ben Jelloun, N.; Febvret, A. Sparing of the dopaminergic neurons containing calbindin-D28k and of the dopaminergic mesocortical projections in weaver mutant mice. Neuroscience 1994, 61, 293–305.
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448.
- Foehring, R.C.; Zhang, X.F.; Lee, J.C.; Callaway, J.C. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. Neurophysiol. 2009, 102, 2326–2333.
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Neurosci. 2019, 13, 43.
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262.
- Hermann, D.; Mezler, M.; Müller, M.K.; Wicke, K.; Gross, G.; Draguhn, A.; Bruehl, C.; Nimmrich, V. Synthetic Aβ oligomers (Aβ1-42 globulomer) modulate presynaptic calcium currents: Prevention of Aβ-induced synaptic deficits by calcium channel blockers. J. Pharmacol. 2013, 702, 44–55.
- Ueda, K.; Shinohara, S.; Yagami, T.; Asakura, K.; Kawasaki, K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: A possible involvement of free radicals. Neurochem. 1997, 68, 265–271.
- Willis, M.; Kaufmann, W.A.; Wietzorrek, G.; Hutter-Paier, B.; Moosmang, S.; Humpel, C.; Hofmann, F.; Windisch, M.; -Günther Knaus, H.; Marksteiner, J. L-type calcium channel CaV 1.2 in transgenic mice overexpressing human AbetaPP751 with the London (V717I) and Swedish (K670M/N671L) mutations. Alzheimers Dis. 2010, 20, 1167–1180.
- Kim, S.; Rhim, H. Effects of amyloid-β peptides on voltage-gated L-type Ca(V)1.2 and Ca(V)1.3 Ca(2+) channels. Cells 2011, 32, 289–294.
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. Med. 2012, 4, a005710.
- Kelly, B.L.; Ferreira, A. Beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons, Biol. Chem. 2006, 281, 28079–28089.
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. Biol. Chem. 2007, 282, 11590–11601.
- Parpura-Gill, A.; Beitz, D.; Uemura, E. The inhibitory effects of beta-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 1997, 754, 65–71.
- Fernández-Tomé, P.; Brera, B.; Arévalo, M.A.; de Ceballos, M.L. Beta-amyloid25-35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Dis. 2004, 15, 580–589.
- Kervern, M.; Angeli, A.; Nicole, O. Selective impairment of some forms of synaptic plasticity by oligomeric amyloid-β peptide in the mouse hippocampus: Implication of extrasynaptic NMDA receptors. Alzheimers Dis. 2012, 32, 183–196.
- Fani, G.; Mannini, B.; Vecchi, G.; Cascella, R.; Cecchi, C.; Dobson, C.M.; Vendruscolo, M.; Chiti, F. Aβ Oligomers Dysregulate Calcium Homeostasis by Mechanosensitive Activation of AMPA and NMDA Receptors. ACS Chem. Neurosci. 2021, 12, 766–781.
- Ferreira, I.L.; Bajouco, L.M.; Mota, S.I.; Auberson, Y.P.; Oliveira, C.R.; Rego, A.C. Amyloid beta peptide 1-42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106.
- Costa, R.O.; Ferreiro, E.; Martins, I.; Santana, I.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M.F. Amyloid β-induced ER stress is enhanced under mitochondrial dysfunction conditions. Aging 2012, 33, 824.e5–824.e16.
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-βoligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Cell. Neurosci. 2015, 26, 191.
- Nomura, I.; Kato, N.; Kita, T.; Takechi, H. Mechanism of impairment of long-term potentiation by amyloid β is independent of NMDA receptors or voltage-dependent calcium channels in hippocampal CA1 pyramidal neurons. Lett. 2005, 391, 1–6.
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; Huang, L.; Xue, P.; Li, B.; Wang, X.; Jin, H.; Wang, J.; Yang, F.; Liu, P.; Zhu, Y.; Sui, S.; Chen, Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Cell Biol. 2012, 14, 177–185.
- Bezprozvanny, I.; Hiesinger, P.R. The synaptic maintenance problem: Membrane recycling, Ca2+ homeostasis and late onset degeneration. Neurodegener. 2013, 8, 23.
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Rev. Drug Discov. 2006, 5, 160–170.
- Francis, P.T. Glutamatergic approaches to the treatment of cognitive and behavioural symptoms of Alzheimer’s disease. Dis. 2008, 5, 241–243.
- Mota, S.I.; Ferreira, I.L.; Pereira, C.; Oliveira, C.R.; Rego, A.C. Amyloid-beta peptide1-42 causes microtubule deregulation through N-methyl-D-aspartate receptors in mature hippocampal cultures. Alzheimer Res. 2012, 9, 844–856.
- Paschen, W. Endoplasmic reticulum: A primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium 2003, 34, 365–383.
- Verkhratsky, A. The endoplasmic reticulum and neuronal calcium signaling. Cell Calcium 2002, 32, 393–404.
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic3xTg-AD mice. Neurosci. 2009, 29, 9458–9470.
- Ferreiro, E.; Oliveira, C.R.; Pereira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. Neurosci. Res. 2004, 76, 872–880.
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M. An endoplasmicreticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Dis. 2006, 23, 669–678.
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-beta-(1-42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. Biol. Chem. 2006, 281, 38440–38447.
- Paula-Lima, A.C.; Adasme, T.; SanMartín, C. Amyloid β-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid. Redox Signal. 2011, 14, 1209–1223.
- Wang, X.; Zheng, W. Ca(2+) homeostasis dysregulation in Alzheimer’s disease: A focus on plasma membrane and cell organelles. FASEB J. 2019, 33, 6697–6712.
- Kelly, J.F.; Furukawa, K.; Barger, S.W.; Rengen, M.R.; Mark, R.J.; Blanc, E.M.; Roth, G.S.; Mattson, M.P. Amyloid beta-peptide disrupts carbachol induced muscarinic cholinergic signal transduction in control neurons. Natl. Acad. Sci. USA 1996, 93, 6753–6758.
- Paschen, W. Dependence of vital cell function on endoplasmic reticulum calcium levels: Implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium 2001, 29, 1–11.
- Pannaccione, A.; Secondo, A.; Molinaro, P.; D’Avanzo, C.; Cantile, M.; Esposito, A.; Boscia, F.; Scorziello, A.; Sirabella, R.; di Renzo, G.; et al. A new concept: Aβ1-42 generates a Hyperfunctional proteolytic NCX3 fragment that delays caspase-12 activation and neuronal death. Neurosci. 2012, 32, 10609–10617.
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008, 59, 190–194.
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Natl. Acad. Sci. USA 1994, 91, 534–538.
- Hirashima, N.; Etcheberrigaray, R.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Govoni, S.; Alkon, D.L. Calcium responses in human fibroblasts: A diagnostic molecular profile for Alzheimer’s disease. Aging 1996, 17, 549–555.
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Rev. Mol. Cell Biol. 2012, 13, 566–578.
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271.
- Goldstein, J.C.; Waterhouse, N.J.; Juin, P.; Evan, G.I.; Green, D.R. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Cell Biol. 2000, 2, 156–162.
- Yao, J.; Irwin; R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Natl. Acad. Sci. USA 2009, 106, 14670–14615.
- Atamna, H.; Frey, W.H. 2nd Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 2007, 7, 297–310.
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. Neurosci. 2013, 33, 17042–17051.
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. Alzheimer’s Dis. 2010, 20, S401–S412.
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. J. Pathol. 2011, 179, 2071–2082.
- Brown, M.R.; Sullivan, P.G.; Geddes, J.W. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. Biol. Chem. 2006, 281, 11658–11668.
- Zervas, M.; Opitz, T.; Edelmann, W.; Wanier, B.; Kucherlapati, R.; Stanton, P.K. Impaired hippocampal long-term potentiation in microtubule-associated protein 1B-deficient mice. Neurosci. Res. 2005, 82, 83–92.
- Fukunaga, K.; Muller, D.; Miyamoto, E. CaM kinase II in long-term potentiation. Int. 1996, 28, 343–358.
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718.
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. Aging enables Ca2+ overload and apoptosis induced by amyloid-beta oligomers in rat hippocampal neurons: Neuroprotection by non-steroidal anti-inflammatory drugs and R-flurbiprofen in aging neurons. Alzheimer’s Dis. 2016, 54, 207–221.
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Dis. 2008, 30, 331–342.
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; Hudry, E.; Bacskai, B.J. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer's disease. Nat Commun. 2020, 11, 2146.
- Supnet, C.; Bezprozvanny, I. Neuronal calcium signaling, mitochondrial dysfunction and Alzheimer’s disease. Alzheimers Dis. 2010, 20 (Suppl. S2), S487–S498.
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in alzheimer disease. Cell Death Dis. 2018, 9, 335.
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094914