Alzheimer’s disease (AD) is the most common age-related neurodegenerative disorder that is characterized by amyloid β-protein deposition in senile plaques, neurofibrillary tangles consisting of abnormally phosphorylated tau protein, and neuronal loss leading to cognitive decline and dementia. Despite extensive research, the exact mechanisms underlying AD remain unknown and effective treatment is not available. Many hypotheses have been proposed to explain AD pathophysiology; however, there is general consensus that the abnormal aggregation of the amyloid β peptide (Aβ) is the initial event triggering a pathogenic cascade of degenerating events in cholinergic neurons. The dysregulation of calcium homeostasis has been studied considerably to clarify the mechanisms of neurodegeneration induced by Aβ. Intracellular calcium acts as a second messenger and plays a key role in the regulation of neuronal functions, such as neural growth and differentiation, action potential, and synaptic plasticity. The calcium hypothesis of AD posits that activation of the amyloidogenic pathway affects neuronal Ca2+ homeostasis and the mechanisms responsible for learning and memory. Aβ can disrupt Ca2+ signaling through several mechanisms, by increasing the influx of Ca2+ from the extracellular space and by activating its release from intracellular stores. Here, we review the different molecular mechanisms and receptors involved in calcium dysregulation in AD and possible therapeutic strategies for improving the treatment.
- protein aggregation
- amyloid β peptide (Aβ)
- toxic oligomers
- amyloid fibrils
- tau protein
- neurodegeneration
- ionic dysregulation
- glutamatergic receptors
- NMDA
- AMPA
1. Calcium Dyshomeostasis in Alzheimer’s Disease
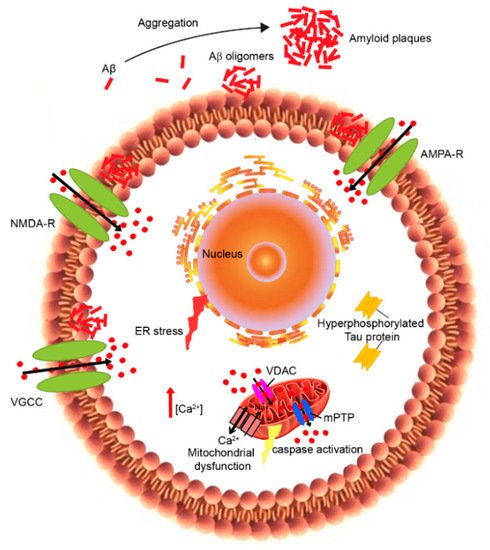
2. Plasma Membrane Calcium Dysregulation
3. Endoplasmic-Reticulum Calcium Dysregulation
4. Mitochondrial Calcium Dyshomeostasis
This entry is adapted from the peer-reviewed paper 10.3390/ijms22094914
References
- Khachaturian, Z.S. Calcium, membranes, aging, and Alzheimer’s disease. Introduction and overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4.
- Landfield, P.W.; Pitler, T.A. Prolonged Ca2+-dependent after hyperpolarizations in hippocampal neurons of aged rats. Science 1984, 226, 1–4.
- Landfield, P.W. Increased calcium current’ hypothesis of brain aging. Neurobiol. Aging 1987, 8, 346–347.
- Verkhratsky, A.; Toescu, E.C. Calcium and neuronal ageing. Trends Neurosci. 1998, 21, 2–7.
- Toescu, E.C.; Verkhratsky, A.; Landfield, P.W. Ca2+ regulation and gene expression in normal brain aging. Trends Neurosci. 2004, 27, 614–620.
- Toescu, E.C.; Verkhratsky, A. The importance of being subtle: Small changes in calcium homeostasis control cognitive decline in normal aging. Aging Cell 2007, 6, 267–273.
- Foster, T.C. Calcium homeostasis and modulation of synaptic plasticity in the aged brain. Aging Cell 2007, 6, 319–325.
- Bezprozvanny, I. Calcium signaling and neurodegenerative diseases. Trends Mol. Med. 2009, 15, 89–100.
- Querfurth, H.W.; Selkoe, D.J. Calcium ionophore increases amyloid β peptide production by cultured cells. Biochemistry 1994, 33, 4550–4561.
- Bezprozvanny, I.; Mattson, M.P. Neuronal calcium mishandling and the pathogenesis of Alzheimer’s disease. Trends Neurosci. 2008, 31, 454–463.
- Etcheberrigaray, R.; Hirashima, N.; Nee, L.; Prince, J.; Govoni, S.; Racchi, M.; Tanzi, R.D.; Alkon, L. Calcium responses in fibroblasts from asymptomatic members of Alzheimer’s disease families. Neurobiol. Dis. 1998, 5, 37–45.
- Latulippe, J.; Lotito, D. A mathematical model for the effects of amyloid beta on intracellular calcium. PLoS ONE 2018, 13, e0202503.
- Kuchibhotla, K.V.; Goldman, S.T.; Lattarulo, C.R.; Wu, H.Y.; Hyman, B.T.; Bacskai, B.J. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron 2008, 59, 214–225.
- Pchitskaya, E.; Popugaeva, E.; Bezprozvanny, I. Calcium signaling and molecular mechanisms underlying neurodegenerative diseases. Cell Calcium 2018, 70, 87–94.
- Demuro, A.; Mina, E.; Kayed, R.; Milton, S.C.; Parker, I.; Glabe, C.G. Calcium dysregulation and membrane disruption as a ubiquitous neurotoxic mechanism of soluble amyloid oligomers. J. Biol. Chem. 2005, 280, 17294–17300.
- Cecchi, C.; Stefani, M. The amyloid-cell membrane system. The interplay between the biophysical features of oligomers/fibrils and cell membrane defines amyloid toxicity. Biophys. Chem. 2013, 182, 30–43.
- Zampagni, M.; Evangelisti, E.; Cascella, R.; Liguri, G.; Becatti, M.; Pensalfini, A.; Uberti, D.; Cenini, G.; Memo, M.; Bagnoli, S.; et al. Lipid rafts are primary mediators of amyloid oxidative attack on plasma membrane. J. Mol. Med. 2010, 88, 597–608.
- Evangelisti, E.; Cecchi, C.; Cascella, R.; Sgromo, C.; Becatti, M.; Dobson, C.M.; Chiti, F.; Stefani, M. Membrane lipid composition and its physicochemical properties define cell vulnerability to aberrant protein oligomers. J. Cell Sci. 2012, 125, 2416–2427.
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Aβ42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225.
- Limbocker, R.; Mannini, B.; Ruggeri, F.S.; Cascella, R.; Xu, C.K.; Perni, M.; Chia, S.; Chen, S.W.; Habchi, J.; Bigi, A.; et al. Trodusquemine displaces protein misfolded oligomers from cell membranes and abrogates their cytotoxicity through a generic mechanism. Commun. Biol. 2020, 3, 435.
- Evangelisti, E.; Cascella, R.; Becatti, M.; Marrazza, G.; Dobson, C.M.; Chiti, F.; Stefani, M.; Cecchi, C. Binding affinity of amyloid oligomers to cellular membranes is a generic indicator of cellular dysfunction in protein misfolding diseases. Sci. Rep. 2016, 6, 32721.
- Evangelisti, E.; Wright, D.; Zampagni, M.; Cascella, R.; Fiorillo, C.; Bagnoli, S.; Relini, A.; Nichino, D.; Scartabelli, T.; Nacmias, B.; et al. Lipid rafts mediate amyloid-induced calcium dyshomeostasis and oxidative stress in Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 143–153.
- Ladiwala, A.R.; Litt, J.; Kane, R.S.; Aucoin, D.S.; Smith, S.O.; Ranjan, S.; Davis, J.; Van Nostrand, W.E.; Tessier, P.M. Conformational differences between two amyloid beta oligomers of similar size and dissimilar toxicity. J. Biol. Chem. 2012, 287, 24765–24773.
- Malchiodi-Albedi, F.; Contrusciere, V.; Raggi, C.; Fecchi, K.; Rainaldi, G.; Paradisi, S.; Matteucci, A.; Santini, M.T.; Sargiacomo, M.; Frank, C.; et al. Lipid raft disruption protects mature neurons against amyloid oligomer toxicity. Biochim. Biophys. Acta 2010, 1802, 406–415.
- Cascella, R.; Evangelisti, E.; Bigi, A.; Becatti, M.; Fiorillo, C.; Stefani, M.; Chiti, F.; Cecchi, C. Soluble oligomers require a ganglioside to trigger neuronal calcium overload. J. Alzheimers Dis. 2017, 60, 923–938.
- Schengrund, C.L. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 2010, 82, 7–17.
- Gunther, E.C.; Strittmatter, S.M. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci. 2012, 15, 1227–1235.
- Hong, S.; Ostaszewski, B.L.; Yang, T.; O’Malley, T.T.; Jin, M.; Yanagisawa, K.; Li, S.; Bartels, T.; Selkoe, D.J. Soluble Abeta oligomers are rapidly sequestered from brain ASF in vivo and bind GM1 ganglioside on cellular membranes. Neuron 2014, 82, 308–319.
- Calamai, M.; Evangelisti, E.; Cascella, R.; Parenti, N.; Cecchi, C.; Stefani, M.; Pavone, F. Single molecule experiments emphasize GM1 as a key player of the different cytotoxicity of structurally distinct Aβ1-42 oligomers. Biochim. Biophys. Acta 2016, 1858, 386–392.
- Fonseca, A.C.R.G.; Moreira, P.I.; Oliveira, C.R.; Cardoso, S.M.; Pinton, P.; Pereira, C.F. Amyloid-beta disrupts calcium and redox homeostasis in brain endothelial cells. Mol. Neurobiol. 2015, 51, 610–622.
- Ranjan, B.; Chong, K.H.; Zheng, J. Composite mathematical modeling of calcium signaling behind neuronal cell death in Alzheimer’s disease. BMC Syst. Biol. 2018, 12, 10.
- Yang, L.; Wang, Z.; Wang, B.; Justice, N.J.; Zheng, H. Amyloid precursor protein regulates Cav1.2 L-type calcium channel levels and function to influence GABAergic short-term plasticity. J. Neurosci. 2009, 29, 15660–15668.
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimers Dis. Parkinsonism 2017, 7, 374.
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid beta protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571.
- Bhatia, R.; Lin, H.; Lal, R. Fresh and globular amyloid beta protein (1–42) induces rapid cellular degeneration: Evidence for Abeta P channel-mediated cellular toxicity. FASEB J. 2000, 14, 1233–1243.
- Kagan, B.L.; Hirakura, Y.; Azimov, R.; Azimova, R.; Lin, M.C. The channel hypothesis of Alzheimer’s disease: Current status. Peptides 2002, 23, 1311–1315.
- Arispe, N.; Diaz, J.C.; Simakova, O. Abeta ion channels. Prospects for treating Alzheimer’s disease with Abeta channel blockers. Biochim. Biophys. Acta 2007, 1768, 1952–1965.
- Sokolov, Y.; Kozak, J.A.; Kayed, R.; Chanturiya, A.; Glabe, C.; Hall, J.E. Soluble amyloid oligomers increase bilayer conductance by altering dielectric structure. J. Gen. Physiol. 2006, 128, 637–647.
- Samad, N.; Ishaq, S.; Bano, S.; Manzoor, N. Calcium Regulation in Alzheimer’s Disease: Mechanistic Understanding. J. Coll. Physicians Surg. Pak. 2017, 27, 566–571.
- Reese, L.C.; Taglialatela, G. A role for calcineurin in Alzheimer’s disease. Curr. Neuropharmacol. 2011, 9, 685–692.
- Yin, Y.; Gao, D.; Wang, Y.; Wang, Z.H.; Wang, X.; Ye, J.; Wu, D.; Fang, L.; Pi, G.; Yang, Y.; et al. Tau accumulation induces synaptic impairment and memory deficit by calcineurin-mediated inactivation of nuclear CaMKIV/CREB signaling. Proc. Natl. Acad. Sci. USA 2016, 113, E3773–E3781.
- Cavallucci, V.; Berretta, N.; Nobili, A.; Nisticò, R.; Mercuri, N.B.; D’Amelio, M. Calcineurin inhibition rescues early synaptic plasticity deficits in a mouse model of Alzheimer’s disease. NeuroMol. Med. 2013, 15, 541–548.
- Li, X.G.; Hong, X.Y.; Wang, Y.L.; Zhang, S.J.; Zhang, J.F.; Li, X.C.; Liu, Y.C.; Sun, D.S.; Feng, Q.; Ye, J.W.; et al. Tau accumulation triggers STAT1-dependent memory deficits by suppressing NMDA receptor expression. EMBO Rep. 2019, 20, e47202.
- Lin, L.; Cao, J.; Yang, S.S.; Fu, Z.Q.; Zeng, P.; Chu, J.; Ning, L.N.; Zhang, T.; Shi, Y.; Tian, Q.; et al. Endoplasmic reticulum stress induces spatial memory deficits by activating GSK-3. J. Cell. Mol. Med. 2018, 22, 3489–3502.
- Liu, Z.C.; Chu, J.; Lin, L.; Song, J.; Ning, L.N.; Luo, H.B.; Yang, S.; Shi, Y.; Wang, Q.; Qu, N.; et al. SIL1 rescued bip elevation-related tau hyperphosphorylation in ER stress. Mol. Neurobiol. 2016, 53, 983–994.
- Zhang, Z.; Li, X.G.; Wang, Z.H.; Song, M.; Yu, S.P.; Kang, S.S.; Liu, X.; Zhang, Z.; Xie, M.; Liu, G.P.; et al. δ-Secretase-cleaved Tau stimulates Aβ production via upregulating STAT1-BACE1 signaling in Alzheimer’s disease. Mol. Psychiatry 2021, 26, 586–603.
- Galla, L.; Redolfi, N.; Pozzan, T.; Pizzo, P.; Greotti, E. Intracellular calcium dysregulation by the Alzheimer’s disease-linked protein presenilin 2. Int. J. Mol. Sci. 2020, 21, 770.
- Gaspar, P.; Ben Jelloun, N.; Febvret, A. Sparing of the dopaminergic neurons containing calbindin-D28k and of the dopaminergic mesocortical projections in weaver mutant mice. Neuroscience 1994, 61, 293–305.
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain 1999, 122, 1437–1448.
- Foehring, R.C.; Zhang, X.F.; Lee, J.C.; Callaway, J.C. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 2009, 102, 2326–2333.
- Liu, J.; Chang, L.; Song, Y.; Li, H.; Wu, Y. The role of NMDA receptors in Alzheimer’s disease. Front. Neurosci. 2019, 13, 43.
- Lombardo, S.; Maskos, U. Role of the nicotinic acetylcholine receptor in Alzheimer’s disease pathology and treatment. Neuropharmacology 2015, 96, 255–262.
- Hermann, D.; Mezler, M.; Müller, M.K.; Wicke, K.; Gross, G.; Draguhn, A.; Bruehl, C.; Nimmrich, V. Synthetic Aβ oligomers (Aβ1-42 globulomer) modulate presynaptic calcium currents: Prevention of Aβ-induced synaptic deficits by calcium channel blockers. Eur. J. Pharmacol. 2013, 702, 44–55.
- Ueda, K.; Shinohara, S.; Yagami, T.; Asakura, K.; Kawasaki, K. Amyloid beta protein potentiates Ca2+ influx through L-type voltage-sensitive Ca2+ channels: A possible involvement of free radicals. J. Neurochem. 1997, 68, 265–271.
- Willis, M.; Kaufmann, W.A.; Wietzorrek, G.; Hutter-Paier, B.; Moosmang, S.; Humpel, C.; Hofmann, F.; Windisch, M.; -Günther Knaus, H.; Marksteiner, J. L-type calcium channel CaV 1.2 in transgenic mice overexpressing human AbetaPP751 with the London (V717I) and Swedish (K670M/N671L) mutations. J. Alzheimers Dis. 2010, 20, 1167–1180.
- Kim, S.; Rhim, H. Effects of amyloid-β peptides on voltage-gated L-type Ca(V)1.2 and Ca(V)1.3 Ca(2+) channels. Mol. Cells 2011, 32, 289–294.
- Lüscher, C.; Malenka, R.C. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb. Perspect. Biol. Med. 2012, 4, a005710.
- Kelly, B.L.; Ferreira, A. Beta-Amyloid-induced dynamin 1 degradation is mediated by N-methyl-D-aspartate receptors in hippocampal neurons. J. Biol. Chem. 2006, 281, 28079–28089.
- De Felice, F.G.; Velasco, P.T.; Lambert, M.P.; Viola, K.; Fernandez, S.J.; Ferreira, S.T.; Klein, W.L. Abeta oligomers induce neuronal oxidative stress through an N-methyl-D-aspartate receptor-dependent mechanism that is blocked by the Alzheimer drug memantine. J. Biol. Chem. 2007, 282, 11590–11601.
- Parpura-Gill, A.; Beitz, D.; Uemura, E. The inhibitory effects of beta-amyloid on glutamate and glucose uptakes by cultured astrocytes. Brain Res. 1997, 754, 65–71.
- Fernández-Tomé, P.; Brera, B.; Arévalo, M.A.; de Ceballos, M.L. Beta-amyloid25–35 inhibits glutamate uptake in cultured neurons and astrocytes: Modulation of uptake as a survival mechanism. Neurobiol. Dis. 2004, 15, 580–589.
- Kervern, M.; Angeli, A.; Nicole, O. Selective impairment of some forms of synaptic plasticity by oligomeric amyloid-β peptide in the mouse hippocampus: Implication of extrasynaptic NMDA receptors. J. Alzheimers Dis. 2012, 32, 183–196.
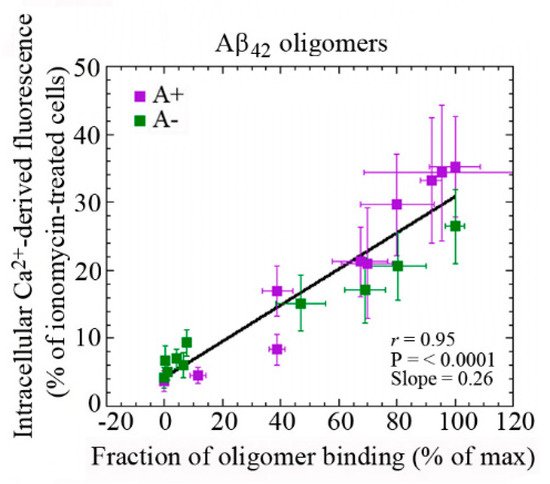
- Fani, G.; Mannini, B.; Vecchi, G.; Cascella, R.; Cecchi, C.; Dobson, C.M.; Vendruscolo, M.; Chiti, F. Aβ Oligomers Dysregulate Calcium Homeostasis by Mechanosensitive Activation of AMPA and NMDA Receptors. ACS Chem. Neurosci. 2021, 12, 766–781.
- Ferreira, I.L.; Bajouco, L.M.; Mota, S.I.; Auberson, Y.P.; Oliveira, C.R.; Rego, A.C. Amyloid beta peptide 1–42 disturbs intracellular calcium homeostasis through activation of GluN2B-containing N-methyl-d-aspartate receptors in cortical cultures. Cell Calcium 2012, 51, 95–106.
- Costa, R.O.; Ferreiro, E.; Martins, I.; Santana, I.; Cardoso, S.M.; Oliveira, C.R.; Pereira, C.M.F. Amyloid β-induced ER stress is enhanced under mitochondrial dysfunction conditions. Neurobiol. Aging 2012, 33, 824.e5–824.e16.
- Ferreira, S.T.; Lourenco, M.V.; Oliveira, M.M.; De Felice, F.G. Soluble amyloid-βoligomers as synaptotoxins leading to cognitive impairment in Alzheimer’s disease. Front. Cell. Neurosci. 2015, 26, 191.
- Nomura, I.; Kato, N.; Kita, T.; Takechi, H. Mechanism of impairment of long-term potentiation by amyloid β is independent of NMDA receptors or voltage-dependent calcium channels in hippocampal CA1 pyramidal neurons. Neurosci. Lett. 2005, 391, 1–6.
- Liu, L.; Feng, D.; Chen, G.; Chen, M.; Zheng, Q.; Song, P.; Ma, Q.; Zhu, C.; Wang, R.; Qi, W.; et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat. Cell Biol. 2012, 14, 177–185.
- Bezprozvanny, I.; Hiesinger, P.R. The synaptic maintenance problem: Membrane recycling, Ca2+ homeostasis and late onset degeneration. Mol. Neurodegener. 2013, 8, 23.
- Lipton, S.A. Paradigm shift in neuroprotection by NMDA receptor blockade: Memantine and beyond. Nat. Rev. Drug Discov. 2006, 5, 160–170.
- Francis, P.T. Glutamatergic approaches to the treatment of cognitive and behavioural symptoms of Alzheimer’s disease. Neurodegener. Dis. 2008, 5, 241–243.
- Mota, S.I.; Ferreira, I.L.; Pereira, C.; Oliveira, C.R.; Rego, A.C. Amyloid-beta peptide1–42 causes microtubule deregulation through N-methyl-D-aspartate receptors in mature hippocampal cultures. Curr. Alzheimer Res. 2012, 9, 844–856.
- Paschen, W. Endoplasmic reticulum: A primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium 2003, 34, 365–383.
- Verkhratsky, A. The endoplasmic reticulum and neuronal calcium signaling. Cell Calcium 2002, 32, 393–404.
- Chakroborty, S.; Goussakov, I.; Miller, M.B.; Stutzmann, G.E. Deviant ryanodine receptor-mediated calcium release resets synaptic homeostasis in presymptomatic3xTg-AD mice. J. Neurosci. 2009, 29, 9458–9470.
- Ferreiro, E.; Oliveira, C.R.; Pereira, C. Involvement of endoplasmic reticulum Ca2+ release through ryanodine and inositol 1,4,5-triphosphate receptors in the neurotoxic effects induced by the amyloid-beta peptide. J. Neurosci. Res. 2004, 76, 872–880.
- Ferreiro, E.; Resende, R.; Costa, R.; Oliveira, C.R.; Pereira, C.M. An endoplasmicreticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiol. Dis. 2006, 23, 669–678.
- Supnet, C.; Grant, J.; Kong, H.; Westaway, D.; Mayne, M. Amyloid-beta-(1–42) increases ryanodine receptor-3 expression and function in neurons of TgCRND8 mice. J. Biol. Chem. 2006, 281, 38440–38447.
- Paula-Lima, A.C.; Adasme, T.; SanMartín, C. Amyloid β-peptide oligomers stimulate RyR-mediated Ca2+ release inducing mitochondrial fragmentation in hippocampal neurons and prevent RyR-mediated dendritic spine remodeling produced by BDNF. Antioxid. Redox Signal. 2011, 14, 1209–1223.
- Wang, X.; Zheng, W. Ca(2+) homeostasis dysregulation in Alzheimer’s disease: A focus on plasma membrane and cell organelles. FASEB J. 2019, 33, 6697–6712.
- Kelly, J.F.; Furukawa, K.; Barger, S.W.; Rengen, M.R.; Mark, R.J.; Blanc, E.M.; Roth, G.S.; Mattson, M.P. Amyloid beta-peptide disrupts carbachol induced muscarinic cholinergic signal transduction in control neurons. Proc. Natl. Acad. Sci. USA 1996, 93, 6753–6758.
- Paschen, W. Dependence of vital cell function on endoplasmic reticulum calcium levels: Implications for the mechanisms underlying neuronal cell injury in different pathological states. Cell Calcium 2001, 29, 1–11.
- Pannaccione, A.; Secondo, A.; Molinaro, P.; D’Avanzo, C.; Cantile, M.; Esposito, A.; Boscia, F.; Scorziello, A.; Sirabella, R.; di Renzo, G.; et al. A new concept: Aβ1-42 generates a Hyperfunctional proteolytic NCX3 fragment that delays caspase-12 activation and neuronal death. J. Neurosci. 2012, 32, 10609–10617.
- Green, K.N.; LaFerla, F.M. Linking calcium to Abeta and Alzheimer’s disease. Neuron 2008, 59, 190–194.
- Ito, E.; Oka, K.; Etcheberrigaray, R.; Nelson, T.J.; McPhie, D.L.; Tofel-Grehl, B.; Gibson, G.E.; Alkon, D.L. Internal Ca2+ mobilization is altered in fibroblasts from patients with Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 534–538.
- Hirashima, N.; Etcheberrigaray, R.; Bergamaschi, S.; Racchi, M.; Battaini, F.; Binetti, G.; Govoni, S.; Alkon, D.L. Calcium responses in human fibroblasts: A diagnostic molecular profile for Alzheimer’s disease. Neurobiol. Aging 1996, 17, 549–555.
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578.
- Görlach, A.; Bertram, K.; Hudecova, S.; Krizanova, O. Calcium and ROS: A mutual interplay. Redox Biol. 2015, 6, 260–271.
- Goldstein, J.C.; Waterhouse, N.J.; Juin, P.; Evan, G.I.; Green, D.R. The coordinate release of cytochrome c during apoptosis is rapid, complete and kinetically invariant. Nat. Cell Biol. 2000, 2, 156–162.
- Yao, J.; Irwin, R.W.; Zhao, L.; Nilsen, J.; Hamilton, R.T.; Brinton, R.D. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2009, 106, 14670–14675.
- Atamna, H.; Frey, W.H. 2nd Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion 2007, 7, 297–310.
- Xie, H.; Guan, J.; Borrelli, L.A.; Xu, J.; Serrano-Pozo, A.; Bacskai, B.J. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J. Neurosci. 2013, 33, 17042–17051.
- Santos, R.X.; Correia, S.C.; Wang, X.; Perry, G.; Smith, M.A.; Moreira, P.I.; Zhu, X. A synergistic dysfunction of mitochondrial fission/fusion dynamics and mitophagy in Alzheimer’s disease. J. Alzheimers Dis. 2010, 20, S401–S412.
- Kopeikina, K.J.; Carlson, G.A.; Pitstick, R.; Ludvigson, A.E.; Peters, A.; Luebke, J.I.; Koffie, R.M.; Frosch, M.P.; Hyman, B.T.; Spires-Jones, T.L. Tau accumulation causes mitochondrial distribution deficits in neurons in a mouse model of tauopathy and in human Alzheimer’s disease brain. Am. J. Pathol. 2011, 179, 2071–2082.
- Brown, M.R.; Sullivan, P.G.; Geddes, J.W. Synaptic mitochondria are more susceptible to Ca2+ overload than nonsynaptic mitochondria. J. Biol. Chem. 2006, 281, 11658–11668.
- Zervas, M.; Opitz, T.; Edelmann, W.; Wanier, B.; Kucherlapati, R.; Stanton, P.K. Impaired hippocampal long-term potentiation in microtubule-associated protein 1B-deficient mice. J. Neurosci. Res. 2005, 82, 83–92.
- Fukunaga, K.; Muller, D.; Miyamoto, E. CaM kinase II in long-term potentiation. Neurochem. Int. 1996, 28, 343–358.
- Sanz-Blasco, S.; Valero, R.A.; Rodriguez-Crespo, I.; Villalobos, C.; Nunez, L. Mitochondrial Ca2+ overload underlies Abeta oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE 2008, 3, e2718.
- Calvo-Rodriguez, M.; Garcia-Durillo, M.; Villalobos, C.; Nunez, L. Aging enables Ca2+ overload and apoptosis induced by amyloid-beta oligomers in rat hippocampal neurons: Neuroprotection by non-steroidal anti-inflammatory drugs and R-flurbiprofen in aging neurons. J. Alzheimers Dis. 2016, 54, 207–221.
- Ferreiro, E.; Oliveira, C.R.; Pereira, C.M. The release of calcium from the endoplasmic reticulum induced by amyloid-beta and prion peptides activates the mitochondrial apoptotic pathway. Neurobiol. Dis. 2008, 30, 331–342.
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146.
- Supnet, C.; Bezprozvanny, I. Neuronal calcium signaling, mitochondrial dysfunction and Alzheimer’s disease. J. Alzheimers Dis. 2010, 20 (Suppl. S2), S487–S498.
- Area-Gomez, E.; De Groof, A.; Bonilla, E.; Montesinos, J.; Tanji, K.; Boldogh, I.; Pon, L.; Schon, E.A. A key role for MAM in mediating mitochondrial dysfunction in alzheimer disease. Cell Death Dis. 2018, 9, 335.
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88.