 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simona Bungău | + 2395 word(s) | 2395 | 2021-04-20 08:18:30 | | | |
| 2 | Nora Tang | Meta information modification | 2395 | 2021-05-12 07:57:36 | | |
Video Upload Options
Neurodegenerative disorders/diseases (NDs) are a chronic debilitating group of heterogeneous diseases, which include loss of neuronal function and structure, leading to neuronal cell death or progressive degeneration. NDs comprise a highly complex etiology that is mainly associated with abnormal protein accumulation, mutated genes, increased reactive oxygen species (ROS), neuroinflammation, mitochondrial dysfunction, apoptosis, elevated endoplasmic reticulum (ER), calcium overload, excitotoxicity, or neuronal destruction in specific regions of the brain. ND is a wide array of neurological disorders that generally affect central nervous system (CNS) neurons, characterized by progressive neuronal dysfunction in the CNS, resulting in deficit of specific functions of the brain (movement, memory, and cognition). These processes are involved in the pathogenesis and progression of NDs, such as Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and Parkinson’s disease.
1. Introduction
ND is a universal terminology for a varied debilitating and often irregular group of disorders categorized by diseases of the dynamic nervous system emerging from neuronal loss and neuronal degradation [1][2]. Apart from neural loss, these NDs are characterized by impaired electro-physiological and neuro-chemical properties in the vulnerable regions of the brain, thus influencing the associated functions [3][4]. Moreover, ND encompasses a wide array of neurological disorders that generally affect CNS neurons, characterized by progressive neuronal dysfunction in the CNS, resulting in deficit of specific functions of the brain (movement, memory, and cognition) [5][6]. NDs comprise a highly complex etiology, mainly associated with abnormal protein accumulation, mutated genes, increased ROS, neuroinflammation, mitochondrial dysfunction, apoptosis, elevated ER stress, calcium overload, excitotoxicity, or neuronal destruction in specific regions of the brain [7][8]. Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and Parkinson’s disease are examples of NDs.
Considered to be second most persisting/recurring ND worldwide with multifaceted etiologies, Parkinson’s disease affects approximately 1–2% of the population [9][10]. An intra-neuronal buildup of protein aggregates and Lewy bodies (consisting of misfolded α-synuclein) and gradual decline of dopaminergic neurons in the substantia nigra pars compacta [11][12] are the two major neuropathological processes involved in Parkinson’s disease. Patients with Parkinson’s suffer from a variety of non-motor symptoms, uncontrollable tremors, and diminished motor function that exacerbate in the later stages of the disease [13][14]. Symptoms include anxiety, dementia, depression, optical delusion, and excessive daytime sleepiness [15]. Several other symptoms may involve cognitive and autonomic dysfunction, olfactory functional loss, and rapid eye movement, which may appear at later stages of this disorder [16].
Alzheimer’s disease is the most common type of ND in the elderly, which worsens with time. Becoming a major healthcare challenge of 21st century, its prevalence continues to rise worldwide. It is the most established cause of dementia characterized by gradual cognitive impairment [17]. Thought to start 20 years or more before, the symptoms arise with brain alterations that remain unnoticeable to the affected person. Individuals after years of brain alterations experience noticeable symptoms (i.e., language problems and memory loss) [17][18][19][20]. The occurrence of symptoms is due to damage or destruction of nerve cells (neurons) in brain regions involved in learning, thinking, and memory [17][21][22]. Therefore, Alzheimer’s disease is characterized by visual-spatial disorders, cognitive functional loss, memory and language impairment, and complications with judgment or analysis. The two proposed fundamental pathogenic mechanisms are the amyloid cascade and the tau hyperphosphorylation [23]. The neuropathological feature includes accumulation of β-amyloid protein, leading to hard plaques interfering with acetylcholine initiating inflammatory processes and affecting synaptic transmission, which may further result in the specific protein (tau) leading to cell demise in Alzheimer’s disease. Therefore, the pairing of microtubules with other tubules of the neurons produces neurofibrillary tangles, resulting in disintegration of tubule and blockade of neurotransmitters, which causes cell death [24][25].
Another progressive ND, amyotrophic lateral sclerosis, is characterized by progressive motor neurodegeneration and death of lower and upper motor neurons in the spinal cord, brain, and brainstem, affecting voluntary muscles and leading to paralysis and respiratory failure and even death [26]. The causative mechanisms underlying amyotrophic lateral sclerosis remain unclear but numerous factors have been included such as excitotoxicity, genetic factors, autoimmune response, protein misfolding and aggregation, oxidative stress, deficits in neurotrophic factors, neurofilament aggregation, environmental factors, mitochondrial dysfunction, and impaired axonal transport [27][28]. This disease is linked with gene mutation that produces superoxide dismutase-1 enzyme. Moreover, this ND is linked with protein inclusions, composed of cytoplasmic trans active response DNA binding protein-43 (TDP-43) in the affected regions of spinal cord and brain [29].
Huntington’s disease is an inherited ND characterized pathologically by diminished functions of gamma-aminobutyric acid and excessive dopaminergic activity in the basal ganglia. Clinically, it is characterized by psychiatric disturbance, abnormal movements, and cognitive deficits [12], being associated with the expansion of the trinucleotide repeat in the Huntington (Htt) gene present at the short arm of chromosome 4. Htt (mutant) proteolysis has been observed to contribute to its pathology, but its role is not yet well defined [30].
An inflammatory, autoimmune, and chronic in nature, multiple sclerosis is a CNS disease characterized by axonal preservation with demyelinated regions. Multiple sclerosis is mostly observed in individuals 20–45 years of age in contrast with other old-age-related NDs [31]. The major driver of pathology in multiple sclerosis is inflammation of CNS. Various factors encourage its development, yet its cause is not well-defined. In MS, the disrupted BBB elicits infiltration of peripheral blood leukocyte, followed by myelin degradation, axonal disruption, and cell loss of neurons. Approximately 2.3 million people are estimated to live with this disease worldwide. Moreover, the prevalence and incidence of this ND is increasing. There are progressive and relapsing–remitting forms of multiple sclerosis; its cause is still not well understood and established [32][33].
2. Mechanisms Involved in Neurodegeneration
Different mechanisms have been implicated in the progression and pathogenesis of NDs, including OS, neuroinflammation, apoptosis, excitotoxicity, and mitochondrial dysfunction [34][35][36][37][38] (Figure 1).
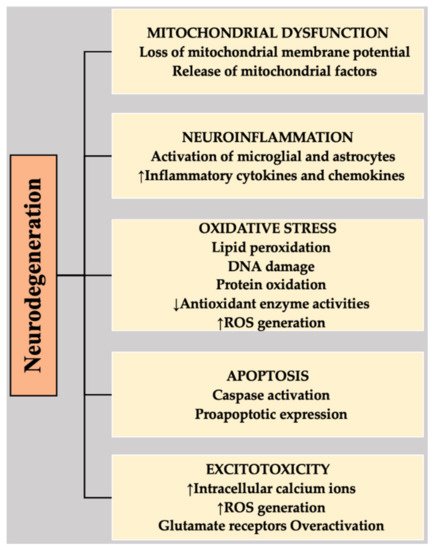
Figure 1. Highlighting the associations of different mechanisms with the neurodegeneration that is involved in NDs. DNA, deoxyribonucleic acid; ROS, reactive oxygen species.
2.1. Apoptosis
Apoptosis facilitates the precisely programmed natural neuronal death that is physiologically essential in neurogenesis during CNS maturation. The process is energy-dependent, which necessitates ATP for signal activation and protein synthesis [39]. Apoptosis is a complicated process, which is triggered by various extrinsic and intrinsic signals. Extrinsic and intrinsic pathways consist of death receptor activation upon ligand binding and production of pro-apoptotic factors in cytosol, respectively. Subsequently, the intrinsic pathway triggers caspase-independent apoptosis or caspase-dependent apoptosis [37][39]. Characterized by chromatin condensation, cell shrinkage, membrane cell death, and DNA fragmentation, neuronal apoptosis is the neuropathological hallmark of NDs. Therefore, abnormality in the regulation of apoptosis or premature apoptosis is involved in the pathogenesis of neurodegeneration, a multifarious process that leads to several NDs, such as Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, Parkinson’s disease, etc. [40].
Various primary brain regions undergo neuronal loss; as diseases progress, these regions can be expanded in several conditions. The most noticeable clinical symptom in patients of Alzheimer’s disease is memory loss, which is associated with neuronal loss in the hippocampal region. As Alzheimer’s disease advances, the cortical/subcortical areas experience neuronal losses that can be extremely severe [41][42]. In the apoptosis context, it has been indicated that executor or initiator caspases are activated in Alzheimer’s disease. Moreover, the levels of extrinsic apoptotic pathway protein have been reported to elevate in the brains of Alzheimer’s disease [43]. In the brains of Parkinson’s disease, it has long been proposed that apoptosis is the preferred pathway for the elimination of dopaminergic neurons in substantia nigra. It has been suggested that nearly every Lewy body neuron is positive for pro-apoptotic process, proposing that heavily burdened neuron with protein aggregates undergo apoptosis [44][45]. In Huntington’s disease, it has been specifically proposed that hyperpolarization of mitochondrial membrane occurs via an mHtt monomer to promote apoptotic neuronal death [46]. In amyotrophic lateral sclerosis, it is TDP-43 that was found to induce the pro-apoptotic proteins expression in a p53-dependent manner. However, various studies suggested that loss of neurons in the brains of amyotrophic lateral sclerosis is a caspase-independent process [47][48].
2.2. Oxidative Stress (OS)
OS is a contributory hallmark in the pathophysiology of common NDs. OS is the imbalance of ROS production and antioxidative defense ability, resulting in cellular damage and DNA repair system damage, which will speed up the neurodegenerative process and progression of NDs [34].
The nervous system is mainly susceptible to OS for the following reasons:
-
lower endogenous antioxidants reserve levels;
-
more oxygen consumption due to higher ATP demand;
-
availability of polyunsaturated lipids that are mainly vulnerable to attack of free reactive species in the neuronal cell membrane;
-
presence of neurotransmitters and excitatory amino acids, the metabolism of which can generate ROS; and
These aforementioned characteristics make neuronal cells a target for the damage caused by ROS. In patients with amyotrophic lateral sclerosis, Aβ neurotoxicity and neuronal degeneration are correlated with the oxidative damage to lipids and proteins, DNA, and RNA [51][52]. In Parkinson’s disease, oxidative damage to proteins and DNA has been reported in the nigro-striatal regions [53][54]. Dopamine oxidation to 6-OHDA (reactive) stipulates an essential endogenous cascade for creating Parkinson’s disease-like condition [55]. Moreover, the overload of transition metals (such as iron) has been implicated in the Lewy body formation process in Parkinson’s [56]. OS-induced damage to DNA, proteins, and lipids and mutations in the SOD-1(Cu/Zn)-encoding gene have been linked with sporadic and familial amyotrophic lateral sclerosis forms [57]. The pathology of multiple sclerosis is not dissimilar when the harmful influence of OS is considered. In addition, reactive species generated by the mononuclear cells and activated microglia play a critical role in the pathogenesis of multiple sclerosis. Declined endogenous antioxidants levels and oxidative damage to mtDNA are related to the axonal injury and demyelination in multiple sclerosis [58]. Exacerbation of lipofuscin and increased occurrence of the oxidative DNA strand breaks is known to be associated with Huntington’s disease [59][60]. It has been found that a substantial increase in 8-OH-dG levels occurs in nDNA in the postmortem tissue of Huntington’s disease subjects [61]. All the aforementioned studies noticeably specify a causal and evidential relationship between OS and neuronal cell demise in NDs.
2.3. Calcium Overload and Excitotoxicity
Excitotoxicity is another process implicated in the pathogenesis of NDs. It is caused by the persistent over-activation of glutamate receptors by excitotoxins or excitatory amino acids in the CNS, leading to neuronal death [34]. Pathologically elevated levels of glutamate and binding of excitotoxins with the glutamate receptors can cause or trigger excitotoxicity by permitting speedy entry of calcium ions (Ca2+) in the cell [62]. In the cell, Ca2+ influx activates numerous Ca2+-dependent enzymes, including endonucleases, protein phosphatases, lipases, endonucleases, xanthine oxidase, proteases, phospholipases, protein kinase, and inducible nitric oxide synthase (iNOS). These enzymes destroy and damage cellular structures such as membrane, cytoskeleton components, and DNA.
The excessive influx of Ca2+ could also render mitochondrial dysfunction, ROS production, OS, and inflammatory responses, ultimately leading to neuronal cell demise [2][63]. Apart from these processes, accumulation of Ca2+ in neurons can also take place via different routes such as activation of receptor-operated Ca2+ channels, voltage-sensitive Ca2+ channels, ATP-dependent Ca2+ channel (P2j receptor), Ca2+ channels coupled to G-protein receptors, and cyclic nucleotide-gated Ca2+ channels [62][64]. An Aβ-induced neuronal cell demise in AD is linked with dysregulation of pathways dependent on Ca2+ [65]. In Parkinson’s disease, intracellular Ca2+ dysregulation can describe the destruction of dopaminergic neurons [66]. Besides, neuronal injury and destabilization in multiple sclerosis are due to over-activation of calpain and Ca2+-dependent protein phosphatases and proteases [67]. The opening of mitochondrial permeability transition pore due to Ca2+ release and mitochondrial Ca2+ overload from the endoplasmic reticulum by mHtt is considered as the process of neuronal demise in Huntington’s disease [68].
2.4. Neuroinflammation
Emerging threads of evidence underscore the role of neuroinflammation in the progression or pathophysiology of NDs. The beginning and consequent increase in neuroinflammation seem to depend on cross-talks among glia, neurons, and immune cells. Macrophages are located in the brain region nearby glial cells that play an essential role in neuroinflammation-mediated NDs. Moreover, activated microglia in the diseased state mediates neuronal injury via release of pro-inflammatory factors. The production of pro-inflammatory factors results in trans-endothelial migration of immune cells across the blood–brain barrier (BBB) [38].
Different processes have been implicated for the actions mediated by microglia that cause neuronal destruction. Phagocytic oxidase (PHOX)-mediated OS-induced neurotoxicity is the foremost mechanism involved here. When PHOX is triggered by inflammatory state, it produces OS by promptly generating high superoxide levels. The inflammatory PHOX activation also stimulates activated microglia leading to production of IL-1β and TNF-α [69][70]. Therefore, activation of PHOX offers an essential link between OS and inflammation [69][71]. Microglia-expressed inducible nitric oxide synthase (iNOS) and astrocytes are upregulated during neuroinflammation, leading to increase production and, thus, promoting neuronal death [34]. However, it should be observed that activation of either NO or NADPH oxidase via iNOS expression alone is insufficient to cause neurotoxicity, but rather their mutual activation triggers the cascade of inflammatory neurodegeneration [69][72]. Moreover, chemokines released by astrocytes exert multifaceted roles in the pathophysiology of chronic NDs including Alzheimer’s disease [34][73].
2.5. Mitochondrial Dysfunction
Considered as the site of cellular respiration and oxidative phosphorylation, mitochondria play an essential role in sustaining a low Ca2+ concentration in the cytosol. Excessive Ca2+ uptake and ROS generation cause the failure of mitochondrial membrane potential and mitochondrial permeability pore opening [74]. The pathophysiology of numerous NDs such as Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and Parkinson’s disease mainly comprises damaged mitochondria [35][74]. Several molecular mechanisms dependent on mitochondria such as generation of ROS, inhibition of mitochondrial electron transport chain (ETC) complexes, dysfunction of the enzymes implicated in the cycle of tricarboxylic acid, perturbations of mitochondrial clearance mechanisms, and impairment of mitochondrial dynamics could promote the pathogenesis of NDs and neuronal injury [35]. In the substantia-nigra, defective ETC (Complex I) is known as the chief cause of sporadic Parkinson’s disease [75]. Mitochondria abnormalities also trigger the neuronal loss and damage in amyotrophic lateral sclerosis and Alzheimer’s disease [76][77]. In multiple sclerosis, neuronal degeneration is also considered to contribute to the irregular activities of ETC (Complexes I and IV) [78]. Several pre-clinical and clinical lines of evidence from Huntington’s disease cases suggest that mitotoxicity plays a critical role in the pathogenesis of neurodegeneration [79]. Owing to complicated cross-talks among mitotoxicity, chronic inflammatory state, and OS, it is not yet well-defined whether damage to mitochondria is the consequence or cause of neuronal dysfunction and damage [80].
References
- Dorszewska, J.; Kozubski, W.; Waleszczyk, W.; Zabel, M.; Ong, K. Neuroplasticity in the Pathology of Neurodegenerative Diseases. Neural Plast. 2020, 2020, 1–2.
- Mehta, A.; Prabhakar, M.; Kumar, P.; Deshmukh, R.; Sharma, P.L. Excitotoxicity: Bridge to various triggers in neurodegenerative disorders. Eur. J. Pharmacol. 2013, 698, 6–18.
- Kovacs, G.G. Concepts and classification of neurodegenerative diseases. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2018; pp. 301–307.
- Mattson, M.P.; Moehl, K.; Ghena, N.; Schmaedick, M.; Cheng, A. Intermittent metabolic switching, neuroplasticity and brain health. Nat. Rev. Neurosci. 2018, 19, 81–94.
- Fan, J.; Dawson, T.M.; Dawson, V.L. Cell Death Mechanisms of Neurodegeneration. In Advances in Neurobiology; Springer International Publishing: Berlin/Heidelberg, Germany, 2017; pp. 403–425.
- Mohd Sairazi, N.S.; Sirajudeen, K.N.S. Natural Products and Their Bioactive Compounds: Neuroprotective Potentials against Neurodegenerative Diseases. Evid.-Based Complement. Altern. Med. 2020, 2020, 1–30.
- Dugger, B.N.; Dickson, D.W. Pathology of Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2017, 9, a028035.
- Makkar, R.; Behl, T.; Bungau, S.; Zengin, G.; Mehta, V.; Kumar, A.; Uddin, M.S.; Ashraf, G.M.; Abdel-Daim, M.M.; Arora, S.; et al. Nutraceuticals in Neurological Disorders. Int. J. Mol. Sci. 2020, 21, 4424.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Yang, W.; Hamilton, J.L.; Kopil, C.; Beck, J.C.; Tanner, C.M.; Albin, R.L.; Ray Dorsey, E.; Dahodwala, N.; Cintina, I.; Hogan, P.; et al. Current and projected future economic burden of Parkinson’s disease in the U.S. NPJ Parkinsons Dis. 2020, 6.
- Behl, T.; Kaur, G.; Fratila, O.; Buhas, C.; Judea-Pusta, C.T.; Negrut, N.; Bustea, C.; Bungau, S. Cross-talks among GBA mutations, glucocerebrosidase, and α-synuclein in GBA-associated Parkinson’s disease and their targeted therapeutic approaches: A comprehensive review. Transl. Neurodegener. 2021, 10.
- Dues, D.J.; Moore, D.J. LRRK2 and Protein Aggregation in Parkinson’s Disease: Insights From Animal Models. Front. Neurosci. 2020, 14.
- Kaur, G.; Behl, T.; Bungau, S.; Kumar, A.; Uddin, M.S.; Mehta, V.; Zengin, G.; Mathew, B.; Shah, M.A.; Arora, S. Dysregulation of the Gut-Brain Axis, Dysbiosis and Influence of Numerous Factors on Gut Microbiota Associated Parkinson’s Disease. Curr. Neuropharmacol. 2020, 19, 233–247.
- Crippa, J.A.S.; Hallak, J.E.C.; Zuardi, A.W.; Guimarães, F.S.; Tumas, V.; dos Santos, R.G. Is cannabidiol the ideal drug to treat non-motor Parkinson’s disease symptoms? Eur. Arch. Psychiatry Clin. Neurosci. 2019, 269, 121–133.
- Chen, H.; Zhao, E.J.; Zhang, W.; Lu, Y.; Liu, R.; Huang, X.; Ciesielski-Jones, A.J.; Justice, M.A.; Cousins, D.S.; Peddada, S. Meta-analyses on prevalence of selected Parkinson’s nonmotor symptoms before and after diagnosis. Transl. Neurodegener. 2015, 4.
- Behl, T.; Kaur, G.; Bungau, S.; Jhanji, R.; Kumar, A.; Mehta, V.; Zengin, G.; Brata, R.; Hassan, S.S.U.; Fratila, O. Distinctive Evidence Involved in the Role of Endocannabinoid Signalling in Parkinson’s Disease: A Perspective on Associated Therapeutic Interventions. Int. J. Mol. Sci. 2020, 21, 6235.
- Kabir, M.T.; Uddin, M.S.; Zaman, S.; Begum, Y.; Ashraf, G.M.; Bin-Jumah, M.N.; Bungau, S.G.; Mousa, S.A.; Abdel-Daim, M.M. Molecular Mechanisms of Metal Toxicity in the Pathogenesis of Alzheimer’s Disease. Mol. Neurobiol. 2020, 58, 1–20.
- Behl, T.; Kaur, I.; Fratila, O.; Brata, R.; Bungau, S. Exploring the Potential of Therapeutic Agents Targeted towards Mitigating the Events Associated with Amyloid-beta Cascade in Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 7443.
- Behl, T.; Kaur, I.; Sehgal, A.; Kumar, A.; Uddin, M.S.; Bungau, S. The Interplay of ABC Transporters in A beta Translocation and Cholesterol Metabolism: Implicating Their Roles in Alzheimer’s Disease. Mol. Neurobiol. 2020, 1–19.
- Chadha, S.; Behl, T.; Sehgal, A.; Kumar, A.; Bungau, S. Exploring the role of mitochondrial proteins as molecular target in Alzheimer’s disease. Mitochondrion 2021, 56, 62–72.
- Shen, H.; Guan, Q.; Zhang, X.; Yuan, C.; Tan, Z.; Zhai, L.; Hao, Y.; Gu, Y.; Han, C. New mechanism of neuroinflammation in Alzheimer’s disease: The activation of NLRP3 inflammasome mediated by gut microbiota. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2020, 100, 109884.
- Uddin, M.S.; Kabir, M.T.; Tewari, D.; Al Mamun, A.; Barreto, G.E.; Bungau, S.G.; Bin-Jumah, M.N.; Abdel-Daim, M.M.; Ashraf, G.M. Emerging Therapeutic Promise of Ketogenic Diet to Attenuate Neuropathological Alterations in Alzheimer’s Disease. Mol. Neurobiol. 2020.
- Borroni, E.; Bohrmann, B.; Grueninger, F.; Prinssen, E.; Nave, S.; Loetscher, H.; Chinta, S.J.; Rajagopalan, S.; Rane, A.; Siddiqui, A.; et al. Sembragiline: A Novel, Selective Monoamine Oxidase Type B Inhibitor for the Treatment of Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2017, 362, 413–423.
- Fan, L.; Mao, C.; Hu, X.; Zhang, S.; Yang, Z.; Hu, Z.; Sun, H.; Fan, Y.; Dong, Y.; Yang, J.; et al. New Insights Into the Pathogenesis of Alzheimer’s Disease. Front. Neurol. 2020, 10.
- Apostolova, L.G. Alzheimer Disease. Contin. Lifelong Learn. Neurol. 2016, 22, 419–434.
- van den Bos, M.A.J.; Geevasinga, N.; Higashihara, M.; Menon, P.; Vucic, S. Pathophysiology and Diagnosis of ALS: Insights from Advances in Neurophysiological Techniques. Int. J. Mol. Sci. 2019, 20, 2818.
- Nishiyama, A.; Warita, H.; Takahashi, T.; Suzuki, N.; Nishiyama, S.; Tano, O.; Akiyama, T.; Watanabe, Y.; Takahashi, K.; Kuroda, H.; et al. Prominent sensory involvement in a case of familial amyotrophic lateral sclerosis carrying the L8V SOD1 mutation. Clin. Neurol. Neurosurg. 2016, 150, 194–196.
- Morgan, S.; Orrell, R.W. Pathogenesis of amyotrophic lateral sclerosis. Br. Med. Bull. 2016, 119, 87–98.
- Prasad, A.; Bharathi, V.; Sivalingam, V.; Girdhar, A.; Patel, B.K. Molecular Mechanisms of TDP-43 Misfolding and Pathology in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2019, 12.
- Jimenez-Sanchez, M.; Licitra, F.; Underwood, B.R.; Rubinsztein, D.C. Huntington’s Disease: Mechanisms of Pathogenesis and Therapeutic Strategies. Cold Spring Harb. Perspect. Med. 2016, 7, a024240.
- Owen Pickrell, W.; Robertson, N.P. Stem cell treatment for multiple sclerosis. J. Neurol. 2016, 263, 2145–2147.
- Hanken, K.; Sander, C.; Qaiser, L.; Schlake, H.P.; Kastrup, A.; Haupts, M.; Eling, P.A.T.M.; Hildebrandt, H. Salivary IL-1ß as an objective measure for fatigue in multiple sclerosis? In Multiple Sclerosis—From Bench to Bedside: Currents Insights into Pathophysiological Concepts and Their Potential Impact on Patients; Rommer, P.S., Weber, M.S., Illes, Z., Eds.; Frontiers Media SA: Lausanne, Switzerland, 2020; pp. 146–154.
- Wagner, C.A.; Roqué, P.J.; Goverman, J.M. Pathogenic T cell cytokines in multiple sclerosis. J. Exp. Med. 2019, 217.
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1583.
- Wu, Y.; Chen, M.; Jiang, J. Mitochondrial dysfunction in neurodegenerative diseases and drug targets via apoptotic signaling. Mitochondrion 2019, 49, 35–45.
- Armada-Moreira, A.; Gomes, J.I.; Pina, C.C.; Savchak, O.K.; Gonçalves-Ribeiro, J.; Rei, N.; Pinto, S.; Morais, T.P.; Martins, R.S.; Ribeiro, F.F.; et al. Going the Extra (Synaptic) Mile: Excitotoxicity as the Road Toward Neurodegenerative Diseases. Front. Cell. Neurosci. 2020, 14.
- Joseph, C.; Mangani, A.S.; Gupta, V.; Chitranshi, N.; Shen, T.; Dheer, Y.; Kb, D.; Mirzaei, M.; You, Y.; Graham, S.L.; et al. Cell Cycle Deficits in Neurodegenerative Disorders: Uncovering Molecular Mechanisms to Drive Innovative Therapeutic Development. Aging Dis. 2020, 11, 946.
- Kwon, H.S.; Koh, S.-H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9.
- Jan, R.; Chaudhry, G.-E.-S. Understanding Apoptosis and Apoptotic Pathways Targeted Cancer Therapeutics. Adv. Pharm. Bull. 2019, 9, 205–218.
- Mattson, M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000, 1, 120–130.
- Martínez-Pinilla, E.; Ordóñez, C.; del Valle, E.; Navarro, A.; Tolivia, J. Regional and Gender Study of Neuronal Density in Brain during Aging and in Alzheimer’s Disease. Front. Aging Neurosci. 2016, 8.
- Guo, H.; Albrecht, S.; Bourdeau, M.; Petzke, T.; Bergeron, C.; LeBlanc, A.C. Active Caspase-6 and Caspase-6-Cleaved Tau in Neuropil Threads, Neuritic Plaques, and Neurofibrillary Tangles of Alzheimer’s Disease. Am. J. Pathol. 2004, 165, 523–531.
- Masliah, E.; Mallory, M.; Alford, M.; Tanaka, S.; Hansen, L.A. Caspase Dependent DNA Fragmentation Might Be Associated with Excitotoxicity in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 1998, 57, 1041–1052.
- Hartmann, A.; Michel, P.P.; Troadec, J.-D.; Mouatt-Prigent, A.; Faucheux, B.A.; Ruberg, M.; Agid, Y.; Hirsch, E.C. Is Bax a mitochondrial mediator in apoptotic death of dopaminergic neurons in Parkinson’s disease? J. Neurochem. 2001, 76, 1785–1793.
- Seidel, K.; Mahlke, J.; Siswanto, S.; Krüger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.T.; Wicht, H.; Korf, H.-W.; et al. The Brainstem Pathologies of Parkinson’s Disease and Dementia with Lewy Bodies. Brain Pathol. 2014, 25, 121–135.
- Grima, J.C.; Daigle, J.G.; Arbez, N.; Cunningham, K.C.; Zhang, K.; Ochaba, J.; Geater, C.; Morozko, E.; Stocksdale, J.; Glatzer, J.C.; et al. Mutant Huntingtin Disrupts the Nuclear Pore Complex. Neuron 2017, 94, 93–107.e106.
- Tsuiji, H.; Inoue, I.; Takeuchi, M.; Furuya, A.; Yamakage, Y.; Watanabe, S.; Koike, M.; Hattori, M.; Yamanaka, K. TDP-43 accelerates age-dependent degeneration of interneurons. Sci. Rep. 2017, 7.
- Vogt, M.A.; Ehsaei, Z.; Knuckles, P.; Higginbottom, A.; Helmbrecht, M.S.; Kunath, T.; Eggan, K.; Williams, L.A.; Shaw, P.J.; Wurst, W.; et al. TDP-43 induces p53-mediated cell death of cortical progenitors and immature neurons. Sci. Rep. 2018, 8.
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778.
- Sharifi-Rad, M.; Anil Kumar, N.V.; Zucca, P.; Varoni, E.M.; Dini, L.; Panzarini, E.; Rajkovic, J.; Tsouh Fokou, P.V.; Azzini, E.; Peluso, I.; et al. Lifestyle, Oxidative Stress, and Antioxidants: Back and Forth in the Pathophysiology of Chronic Diseases. Front. Physiol. 2020, 11.
- Cheignon, C.; Tomas, M.; Bonnefont-Rousselot, D.; Faller, P.; Hureau, C.; Collin, F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018, 14, 450–464.
- Butterfield, D.A.; Boyd-Kimball, D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018, 62, 1345–1367.
- Kikuchi, A.; Takeda, A.; Onodera, H.; Kimpara, T.; Hisanaga, K.; Sato, N.; Nunomura, A.; Castellani, R.J.; Perry, G.; Smith, M.A.; et al. Systemic Increase of Oxidative Nucleic Acid Damage in Parkinson’s Disease and Multiple System Atrophy. Neurobiol. Dis. 2002, 9, 244–248.
- Zhang, J.; Perry, G.; Smith, M.A.; Robertson, D.; Olson, S.J.; Graham, D.G.; Montine, T.J. Parkinson’s Disease Is Associated with Oxidative Damage to Cytoplasmic DNA and RNA in Substantia Nigra Neurons. Am. J. Pathol. 1999, 154, 1423–1429.
- Soto-Otero, R.; Méndez-Álvarez, E.; Hermida-Ameijeiras, Á.; Muñoz-Patiño, A.M.; Labandeira-Garcia, J.L. Autoxidation and Neurotoxicity of 6-Hydroxydopamine in the Presence of Some Antioxidants. J. Neurochem. 2002, 74, 1605–1612.
- Mochizuki, H.; Choong, C.-J.; Baba, K. Parkinson’s disease and iron. J. Neural Transm. 2020, 127, 181–187.
- Cunha-Oliveira, T.; Montezinho, L.; Mendes, C.; Firuzi, O.; Saso, L.; Oliveira, P.J.; Silva, F.S.G. Oxidative Stress in Amyotrophic Lateral Sclerosis: Pathophysiology and Opportunities for Pharmacological Intervention. Oxid. Med. Cell. Longev. 2020, 2020, 5021694.
- Ohl, K.; Tenbrock, K.; Kipp, M. Oxidative stress in multiple sclerosis: Central and peripheral mode of action. Exp. Neurol. 2016, 277, 58–67.
- Kumar, A.; Ratan, R.R. Oxidative Stress and Huntington’s Disease: The Good, The Bad, and The Ugly. J. Huntingt. Dis. 2016, 5, 217–237.
- Tellez-Nagel, I.; Johnson, A.B.; Terry, R.D. Studies on brain biopsies of patients with huntington’s chorea. J. Neuropathol. Exp. Neurol. 1974, 33, 308–332.
- Tang, Q.; Liu, H.; Shi, X.-J.; Cheng, Y. Blood Oxidative Stress Marker Aberrations in Patients with Huntington’s Disease: A Meta-Analysis Study. Oxid. Med. Cell. Longev. 2020, 2020, 9187195.
- Wojda, U.; Salinska, E.; Kuznicki, J. Calcium ions in neuronal degeneration. IUBMB Life 2008, 60, 575–590.
- Fakhoury, M. Role of Immunity and Inflammation in the Pathophysiology of Neurodegenerative Diseases. Neurodegener. Dis. 2015, 15, 63–69.
- Frankel, E.B.; Kurshan, P.T. All Talk, No Assembly: Voltage-Gated Calcium Channels Do Not Mediate Active Zone Formation. Neuron 2020, 107, 593–594.
- Wang, Y.; Shi, Y.; Wei, H. Calcium Dysregulation in Alzheimer’s Disease: A Target for New Drug Development. J. Alzheimers Dis. Parkinsonism 2017, 7.
- Zaichick, S.V.; McGrath, K.M.; Caraveo, G. The role of Ca2+ signaling in Parkinson’s disease. Dis. Models Mech. 2017, 10, 519–535.
- Vosler, P.S.; Brennan, C.S.; Chen, J. Calpain-Mediated Signaling Mechanisms in Neuronal Injury and Neurodegeneration. Mol. Neurobiol. 2008, 38, 78–100.
- Estrada Sánchez, A.M.; Mejía-Toiber, J.; Massieu, L. Excitotoxic Neuronal Death and the Pathogenesis of Huntington’s Disease. Arch. Med. Res. 2008, 39, 265–276.
- Bal-Price, A.; Matthias, A.; Brown, G.C. Stimulation of the NADPH oxidase in activated rat microglia removes nitric oxide but induces peroxynitrite production. J. Neurochem. 2002, 80, 73–80.
- Mander, P.K.; Jekabsone, A.; Brown, G.C. Microglia Proliferation Is Regulated by Hydrogen Peroxide from NADPH Oxidase. J. Immunol. 2006, 176, 1046–1052.
- Segal, A.W. The function of the NADPH oxidase of phagocytes and its relationship to other NOXs in plants, invertebrates, and mammals. Int. J. Biochem. Cell Biol. 2008, 40, 604–618.
- Brown, G.C. Mechanisms of inflammatory neurodegeneration: INOS and NADPH oxidase. Biochem. Soc. Trans. 2007, 35, 1119–1121.
- Jorda, A.; Campos-Campos, J.; Iradi, A.; Aldasoro, M.; Aldasoro, C.; Vila, J.M.; Valles, S.L. The Role of Chemokines in Alzheimer’s Disease. Endocr. Metab. Immune Disord. Drug Targets 2020, 20, 1383–1390.
- Kim, H.-R.; Won, S.J.; Fabian, C.; Kang, M.-G.; Szardenings, M.; Shin, M.-G. Mitochondrial DNA Aberrations and Pathophysiological Implications in Hematopoietic Diseases, Chronic Inflammatory Diseases, and Cancers. Ann. Lab. Med. 2015, 35, 1–14.
- Scorziello, A.; Borzacchiello, D.; Sisalli, M.J.; Di Martino, R.; Morelli, M.; Feliciello, A. Mitochondrial Homeostasis and Signaling in Parkinson’s Disease. Front. Aging Neurosci. 2020, 12.
- Wang, W.; Zhao, F.; Ma, X.; Perry, G.; Zhu, X. Mitochondria dysfunction in the pathogenesis of Alzheimer’s disease: Recent advances. Mol. Neurodegener. 2020, 15.
- Calió, M.L.; Henriques, E.; Siena, A.; Bertoncini, C.R.A.; Gil-Mohapel, J.; Rosenstock, T.R. Mitochondrial Dysfunction, Neurogenesis, and Epigenetics: Putative Implications for Amyotrophic Lateral Sclerosis Neurodegeneration and Treatment. Front. Neurosci. 2020, 14.
- Licht-Mayer, S.; Campbell, G.R.; Canizares, M.; Mehta, A.R.; Gane, A.B.; McGill, K.; Ghosh, A.; Fullerton, A.; Menezes, N.; Dean, J.; et al. Enhanced axonal response of mitochondria to demyelination offers neuroprotection: Implications for multiple sclerosis. Acta Neuropathol. 2020, 140, 143–167.
- Lopes, C.; Tang, Y.; Anjo, S.I.; Manadas, B.; Onofre, I.; de Almeida, L.P.; Daley, G.Q.; Schlaeger, T.M.; Rego, A.C.C. Mitochondrial and Redox Modifications in Huntington Disease Induced Pluripotent Stem Cells Rescued by CRISPR/Cas9 CAGs Targeting. Front. Cell Dev. Biol. 2020, 8.
- Chen, L.; Zhang, Z.; Nie, S. Targeting prion-like protein spreading in neurodegenerative diseases. Neural Regen. Res. 2018, 13, 1875.


