1. Introduction
ND is a universal terminology for a varied debilitating and often irregular group of disorders categorized by diseases of the dynamic nervous system emerging from neuronal loss and neuronal degradation [
14,
15]. Apart from neural loss, these NDs are characterized by impaired electro-physiological and neuro-chemical properties in the vulnerable regions of the brain, thus influencing the associated functions [
16,
17]. Moreover, ND encompasses a wide array of neurological disorders that generally affect CNS neurons, characterized by progressive neuronal dysfunction in the CNS, resulting in deficit of specific functions of the brain (movement, memory, and cognition) [
5,
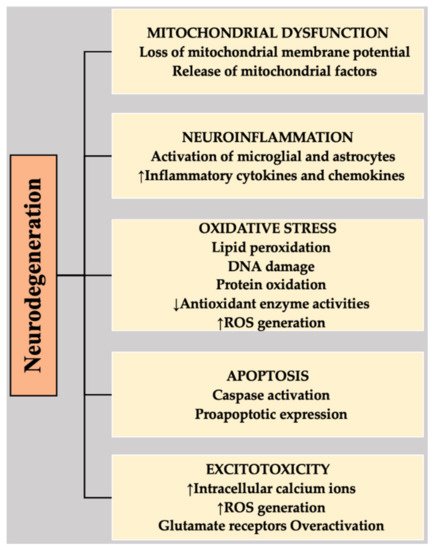
6]. NDs comprise a highly complex etiology, mainly associated with abnormal protein accumulation, mutated genes, increased ROS, neuroinflammation, mitochondrial dysfunction, apoptosis, elevated ER stress, calcium overload, excitotoxicity, or neuronal destruction in specific regions of the brain [
1,
4]. Huntington’s disease, Alzheimer’s disease, amyotrophic lateral sclerosis, multiple sclerosis, and Parkinson’s disease are examples of NDs.
Considered to be second most persisting/recurring ND worldwide with multifaceted etiologies, Parkinson’s disease affects approximately 1–2% of the population [
18,
19]. An intra-neuronal buildup of protein aggregates and Lewy bodies (consisting of misfolded α-synuclein) and gradual decline of dopaminergic neurons in the substantia nigra pars compacta [
20,
21] are the two major neuropathological processes involved in Parkinson’s disease. Patients with Parkinson’s suffer from a variety of non-motor symptoms, uncontrollable tremors, and diminished motor function that exacerbate in the later stages of the disease [
22,
23]. Symptoms include anxiety, dementia, depression, optical delusion, and excessive daytime sleepiness [
24]. Several other symptoms may involve cognitive and autonomic dysfunction, olfactory functional loss, and rapid eye movement, which may appear at later stages of this disorder [
25].
Alzheimer’s disease is the most common type of ND in the elderly, which worsens with time. Becoming a major healthcare challenge of 21st century, its prevalence continues to rise worldwide. It is the most established cause of dementia characterized by gradual cognitive impairment [
26]. Thought to start 20 years or more before, the symptoms arise with brain alterations that remain unnoticeable to the affected person. Individuals after years of brain alterations experience noticeable symptoms (i.e., language problems and memory loss) [
26,
27,
28,
29]. The occurrence of symptoms is due to damage or destruction of nerve cells (neurons) in brain regions involved in learning, thinking, and memory [
26,
30,
31]. Therefore, Alzheimer’s disease is characterized by visual-spatial disorders, cognitive functional loss, memory and language impairment, and complications with judgment or analysis. The two proposed fundamental pathogenic mechanisms are the amyloid cascade and the tau hyperphosphorylation [
32]. The neuropathological feature includes accumulation of β-amyloid protein, leading to hard plaques interfering with acetylcholine initiating inflammatory processes and affecting synaptic transmission, which may further result in the specific protein (tau) leading to cell demise in Alzheimer’s disease. Therefore, the pairing of microtubules with other tubules of the neurons produces neurofibrillary tangles, resulting in disintegration of tubule and blockade of neurotransmitters, which causes cell death [
33,
34].
Another progressive ND, amyotrophic lateral sclerosis, is characterized by progressive motor neurodegeneration and death of lower and upper motor neurons in the spinal cord, brain, and brainstem, affecting voluntary muscles and leading to paralysis and respiratory failure and even death [
35]. The causative mechanisms underlying amyotrophic lateral sclerosis remain unclear but numerous factors have been included such as excitotoxicity, genetic factors, autoimmune response, protein misfolding and aggregation, oxidative stress, deficits in neurotrophic factors, neurofilament aggregation, environmental factors, mitochondrial dysfunction, and impaired axonal transport [
36,
37]. This disease is linked with gene mutation that produces superoxide dismutase-1 enzyme. Moreover, this ND is linked with protein inclusions, composed of cytoplasmic trans active response DNA binding protein-43 (TDP-43) in the affected regions of spinal cord and brain [
38].
Huntington’s disease is an inherited ND characterized pathologically by diminished functions of gamma-aminobutyric acid and excessive dopaminergic activity in the basal ganglia. Clinically, it is characterized by psychiatric disturbance, abnormal movements, and cognitive deficits [
21], being associated with the expansion of the trinucleotide repeat in the Huntington (Htt) gene present at the short arm of chromosome 4. Htt (mutant) proteolysis has been observed to contribute to its pathology, but its role is not yet well defined [
39].
An inflammatory, autoimmune, and chronic in nature, multiple sclerosis is a CNS disease characterized by axonal preservation with demyelinated regions. Multiple sclerosis is mostly observed in individuals 20–45 years of age in contrast with other old-age-related NDs [
40]. The major driver of pathology in multiple sclerosis is inflammation of CNS. Various factors encourage its development, yet its cause is not well-defined. In MS, the disrupted BBB elicits infiltration of peripheral blood leukocyte, followed by myelin degradation, axonal disruption, and cell loss of neurons. Approximately 2.3 million people are estimated to live with this disease worldwide. Moreover, the prevalence and incidence of this ND is increasing. There are progressive and relapsing–remitting forms of multiple sclerosis; its cause is still not well understood and established [
41,
42].