Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Karl Kunzelmann | + 2399 word(s) | 2399 | 2021-06-10 10:18:11 | | | |
| 2 | Rita Xu | -6 word(s) | 2393 | 2021-06-22 11:52:54 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Kunzelmann, K. Phenotype in Polycystic Kidney Disease. Encyclopedia. Available online: https://encyclopedia.pub/entry/11135 (accessed on 08 February 2026).
Kunzelmann K. Phenotype in Polycystic Kidney Disease. Encyclopedia. Available at: https://encyclopedia.pub/entry/11135. Accessed February 08, 2026.
Kunzelmann, Karl. "Phenotype in Polycystic Kidney Disease" Encyclopedia, https://encyclopedia.pub/entry/11135 (accessed February 08, 2026).
Kunzelmann, K. (2021, June 22). Phenotype in Polycystic Kidney Disease. In Encyclopedia. https://encyclopedia.pub/entry/11135
Kunzelmann, Karl. "Phenotype in Polycystic Kidney Disease." Encyclopedia. Web. 22 June, 2021.
Copy Citation
Autosomal dominant polycystic kidney disease (ADPKD) is caused by loss of function of PKD1 (polycystin 1) or PKD2 (polycystin 2). The Ca2+-activated Cl− channel TMEM16A has a central role in ADPKD. Expression and function of TMEM16A is upregulated in ADPKD which causes enhanced intracellular Ca2+ signaling, cell proliferation, and ion secretion.
TMEM16A
ADPKD
polycystic kidneys
androgen
estrogen
1. Introduction
Male gender is a risk factor for progression of autosomal-dominant polycystic kidney disease (ADPKD) [1][2]. Affected men demonstrate faster loss of renal function and earlier onset of end stage renal disease, when compared to women [3]. Orchiectomy led to reduced renal size and cyst volume density, indicating attenuation of renal disease. In contrast, testosterone substitution was shown to antagonize the protective effect of gonadal ablation [4]. Moreover, in females, testosterone increased kidney size and cyst growth, clearly identifying androgens as a progression factor. On the other hand, estrogens have been proposed as protective hormones [5]. These results have been confirmed in additional experiments with rats [6][7]. Thus both, androgens and estrogens have an impact on cyst growth and disease progression in ADPKD.
Evidence has been provided for a crucial role of two chloride ion channels in the pathology of ADPKD, namely protein kinase A-regulated cystic fibrosis transmembrane conductance regulator (CFTR) and the Ca2+-activated Cl− channel transmembrane 16A (TMEM16A). Support for a pro-secretory role of CFTR in ADPKD came from a number of in vitro studies [8][9][10][11], ex vivo experiments in embryonic renal cysts in metanephric organ culture [12][13], and observations in vivo in animals and humans [12][14][15]. In contrast, another in vivo study could not confirm a protective effect of missing CFTR-function in cystic fibrosis for ADPKD [16].
Subsequent work identified a contribution of purinergic Ca2+ signaling to ADPKD [17][18][19]. In a series of studies in vitro, in metanephric renal organ cultures, and in mice in vivo, we demonstrated the essential contribution of TMEM16A to renal cyst formation in ADPKD [20][21][22][23][24]. We showed that knockout of Tmem16a or inhibition of TMEM16A in vivo by the FDA-approved drugs such as niclosamide, benzbromarone, and the TMEM16A-specific inhibitor Ani9 largely reduced cyst enlargement and abnormal cyst cell proliferation. Based on these results, we proposed a novel therapeutic concept for the treatment of ADPKD, based on the inhibition of TMEM16A.
2. Male Mice Lacking Expression of Pkd1 Show a Larger Number of Renal Cysts, But Similar Levels of Expression of TMEM16A
The KspCreERT2;Pkd1lox;lox system was used to obtain a tamoxifen inducible tubule-specific knockout of Pkd1, as described previously [24][25]. Knockdown of Pkd1 in male and female mice was validated by RT-PCR (Figure 1A,B). Ten weeks old Pkd1−/− mice demonstrated multiple renal cysts, which were more pronounced in males when compared to females (Figure 1C,D).
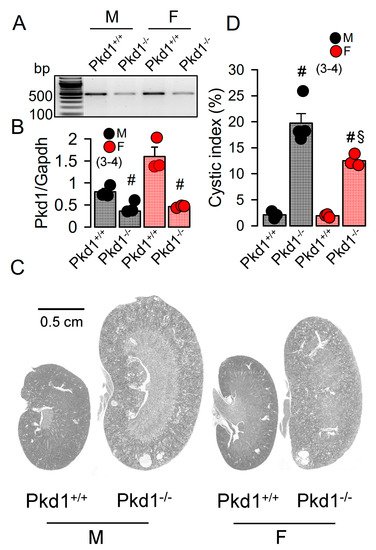
Figure 1. Renal cysts in male and female PKD1-knockout mice. (A,B) RT-PCR of Pkd1 in renal primary epithelial cells from male (M) and female (F) Pkd1+/+ and Pkd1−/− mice, and semiquantitative analysis of expression. (C) HE staining of whole kidneys and analysis of renal cysts by stitching microscopy. Male Pkd1−/− kidneys had larger sizes when compared to kidneys from female Pkd1−/− mice. (D) Cystic index in male and female Pkd1+/+ and Pkd1−/− kidneys. Mean ± SEM (number of animals in each series). # significant difference when compared to Pkd1+/+ (p < 0.05; unpaired t-test). § significant difference when compared to male (p < 0.05; ANOVA with Tukey’s post-hoc test).
In our previous study we reported a crucial role of the Ca2+-activated Cl− channel TMEM16A for the development of polycystic kidneys in Pkd1−/− mice and in additional in vitro models for ADPKD [20][23][24]. We therefore expected to find higher levels of TMEM16A-expression in kidneys from male Pkd1−/− mice. However, both Western blotting of TMEM16A from renal lysates, as well as immunohistochemistry suggested similar levels of TMEM16A-expression in kidneys from male and female Pkd1−/− mice (Figure 2). Similarly, mRNA-expression for the epithelial Cl− channel cystic fibrosis transmembrane conductance regulator (CFTR) was not different between males and females.
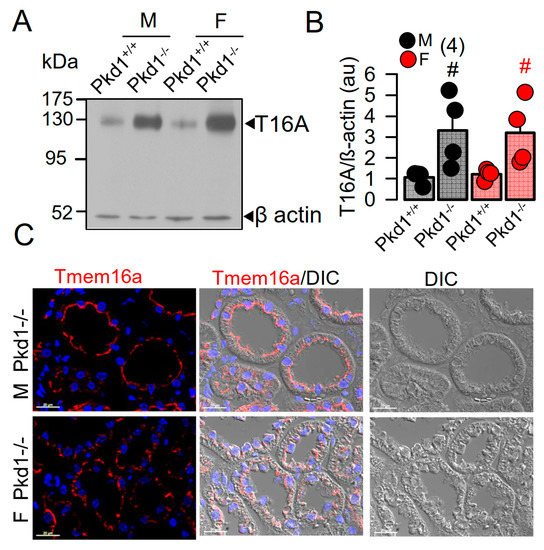
Figure 2. Expression of TMEM16A in male and female PKD1−/− mice. (A,B) Western blotting of TMEM16A-expression in primary renal epithelial cells from kidneys of male and female Pkd1+/+ and Pkd1−/− mice. (C) Immunocytochemistry of Tmem16a in kidneys from male and female Pkd1−/− mice. Bar = 20 µm. Representative images from three mice each. Mean ± SEM (number of animals in each series). # significant difference when compared to Pkd1+/+ (p < 0.05; unpaired t-test).
3. Cell Proliferation and Basal Ca2+ Levels Are More Enhanced in Renal Epithelial Cells from Pkd1−/− Males Than Pkd1−/− Females
Cell proliferation is enhanced in kidneys from Pkd1−/− mice [24]. We compared the cell proliferation in males and females using Ki-67 staining. The results show that cell proliferation is enhanced in kidneys of both male and female Pkd1−/− mice, however, proliferation was more enhanced in males (Figure 3A,B). Cellular Ca2+ levels are intimately related with cell proliferation, and have been shown to be augmented in renal epithelial cells from mice lacking expression of Pkd1, which leads to enhanced expression of TMEM16A [23][24][26]. We therefore compared the basal Ca2+ levels in primary renal epithelial cells from male and female mice. Remarkably, knockout of Pkd1 caused higher basal Ca2+ levels in renal epithelial cells from males, but not in cells from female kidneys (Figure 3C,D). These results suggest a contribution of enhanced intracellular Ca2+ levels to augmented cell proliferation and cyst development in male kidneys.
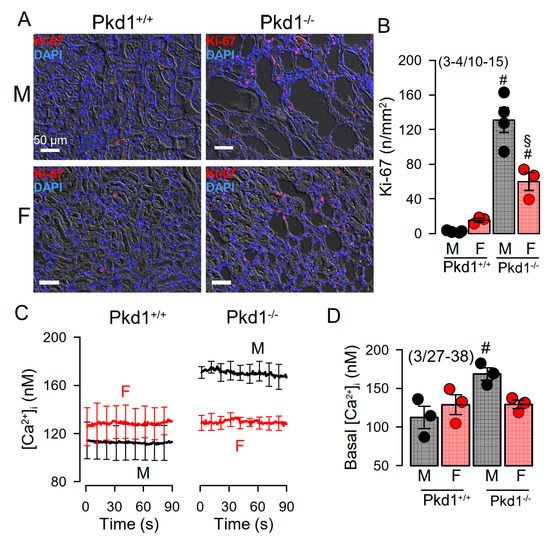
Figure 3. Cell proliferation and intracellular Ca2+ concentrations in renal epithelial cells from Pkd1+/+ and Pkd1−/− mice. (A) Analysis of tubular epithelial cell proliferation in kidney sections from male and female Pkd1+/+ and Pkd1−/− mice as indicates by Ki-67 staining. Bars = 50 µm. (B) Summary of Ki-67 positive cells/mm2 tissue area in kidneys from male and female Pkd1+/+ and Pkd1−/− mice. Bars = 50 µm. (C,D) Intracellular basal Ca2+ concentrations ([Ca2+]i) as assessed by Fura2 indicates higher basal [Ca2+]iI in male Pkd1−/− mice. Mean ± SEM (number of animals/number of experiments in each series). # Significant difference when compared to Pkd1+/+ (p < 0.05; unpaired t-test). § Significant difference when compared to male (p < 0.05; ANOVA and Tukey’s post-hoc test).
We further compared the whole cell currents measured in primary renal epithelial cells isolated from male and female kidneys. Primary cells were isolated from male and female Pkd1+/+ and Pkd1−/− mice. As reported previously, in cells isolated from male or female Pkd1+/+ animals, we found little activation of TMEM16A currents by increase of intracellular Ca2+, using the purinergic ligand ATP (Figure 4A,B, left panels). In contrast, cells isolated from Pkd1−/− animals showed large ATP-activated whole cell currents. We noticed that the basal current in male Pkd1−/− cells was significantly larger than that measured in female Pkd1−/− cells (Figure 4A,B, right panels). This may be explained by the fact that male Pkd1−/− cells showed a higher basal intracellular Ca2+ concentration (Figure 3C,D). We also examined the whole cell currents activated by IBMX and forskolin (IF), which both increase intracellular cAMP. No activation of whole cell currents was observed in Pkd1+/+ cells, while a small but significant current was activated in male Pkd1−/− cells (Figure 4C,D). Patch clamp ion current data were supported by additional iodide quenching data, which also indicated a larger halide permeability in male when compared to female Pkd1−/− cells. Taken together, higher basal Ca2+ concentrations, enhanced proliferation and larger basal Cl− currents may explain the more pronounced cystic phenotype in male Pkd1−/− kidneys.
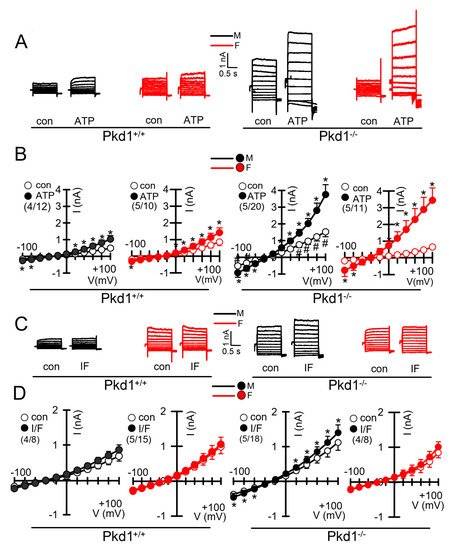
Figure 4. Whole cell patch clamp experiments in primary renal epithelial cells from male and female Pkd1+/+ and Pkd1−/− mice. (A) Original whole cell overlay currents of basal and ATP (50 µM)—activated currents in male and female Pkd1+/+ and Pkd1−/− mice. (B) Summary i/v curves of basal and ATP-activated currents indicating larger ATP-activated currents in Pkd1−/− mice and larger basal currents in male Pkd1−/− mice. (C) Original overlay currents of basal and IF (100 µM IBMX and 2 µM forskolin)—activated currents in male and female Pkd1+/+ and Pkd1−/− mice. (D) Summary i/v curves of basal and IF-activated currents indicating small but significant IF-activated currents in male Pkd1−/− mice. Mean ± SEM (number of animals/number of experiments in each series). * Significant activation by ATP and IF, respectively (p < 0.05; paired t-test). # Significant difference compared to other basal currents (p < 0.05; ANOVA and Tukey’s post-hoc test).
4. Testosterone Augments ATP-Induced Whole Cell Currents in Female Pkd1+/+ Cells
Androgens have been implicated in the severity of the disease phenotype in ADPKD [27]. We therefore examined the impact of dihydrotestosterone (DHT) on the ion currents in primary renal epithelial cells, isolated from female Pkd+/+ mice. Expression of androgen receptors in primary renal epithelial cells was determined by RT-PCR (Figure 5A,B). Moreover, different types of estrogen receptors were found to be expressed in these cell (Figure 5C). As expected, ATP-activated whole cell currents were moderate in control cells, but were significantly enhanced in cells treated with DHT. In contrast, in cells treated with the inhibitor of androgen receptors, cyproterone acetate (CA), no whole cell currents could be activated by ATP (Figure 5D,E).
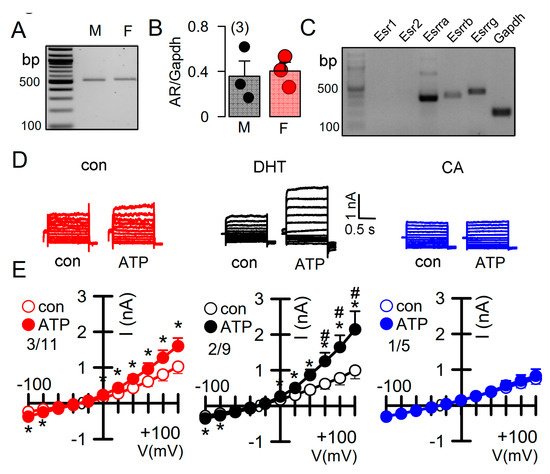
Figure 5. Effect of dihydrotestosterone on whole cell currents in primary renal epithelial cells from female Pkd1+/+ mice. (A,B) RT-PCR analysis of expression of the androgen receptor (AR) in primary renal epithelial cells from male and female kidneys. (C) Expression of estrogen receptors in primary renal epithelial cells from female Pkd1+/+ mice. (D,E) Whole cell current overlays and corresponding I/V-curves from primary renal epithelial cells of female Pkd1+/+ mice. ATP (50 µM) -activated currents were augmented in cells-incubated with dihydrotestosterone (DHT; 10 µM, 24 h), but were absent in cyproterone acetate (CA, 10 µM, 24 h) incubated cells. Mean ± SEM (number of animals/number of experiments in each series). * Significant activation by ATP (p < 0.05; paired t-test). # Significant difference when compared to con (no DHT) (p < 0.05; ANOVA and Tukey’s post-hoc test).
DHT-dependent regulation of Ca2+-activated Cl− currents was further examined in mCCDcl1 mouse renal cortical collecting duct cells [28]. These cells express TMEM16A, CFTR, and receptors for testosterone and estrogen (Figure 6A,B). However, the main estrogene receptors Esr1 and Esr2 are not expressed in renal epithelial cells, suggesting that estrogen-dependent regulation of protein expression is not dominant in kidney.
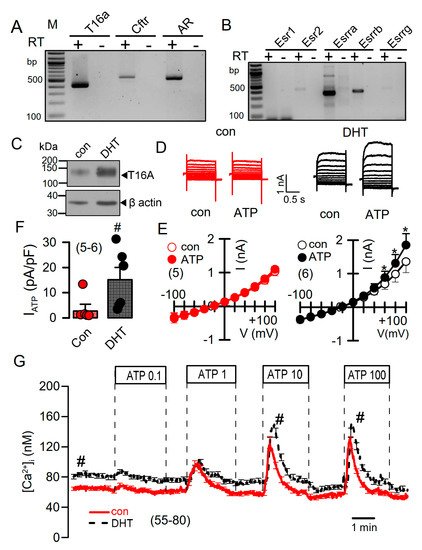
Figure 6. Effect of dihydrotestosterone on whole cell currents and intracellular Ca2+ concentrations in mouse mCCDcl1 cortical collecting duct cells. (A,B) RT-PCR analysis of expression of Tmem16a, Cftr, androgen receptors (A), and estrogen receptors (B) in Pkd1+/+ mCCDcl1 mouse collecting duct cells. (C) Western blot from mCCDcl1 cells, indicating upregulation of Tmem16a-expression by DHT. (D,E) Whole cell current overlays (D), corresponding I/V-curves (E), and summary of ATP (0.1 µM) -activated whole cell currents (F) from mCCDcl1 cells, indicating larger basal and ATP-activated currents in DHT (10 µM; 24 h) treated cells. Current densities (pA/pF) were assessed at the clamp voltage of +100 mV. (G) Basal and ATP (0.1–100 µM)–induced increase in intracellular Ca2+ concentration in control (con) and DHT-incubated mCCDcl1 cells. Mean ± SEM (number of experiments). * Significant activation by ATP (p < 0.05; paired t-test). # Significant difference compared to con (p < 0.05; unpaired t-test).
Treatment with DHT-enhanced expression of TMEM16A in mCCDcl1 cells (Figure 6C). In these cells, ATP concentrations as low as 0.1 µM stimulated a whole cell current that was not observed in the absence of DHT (Figure 6D–F). Upregulation of ATP-activated whole cell currents by DHT was also observed in mouse M1 collecting duct cells, which also showed a slight but detectable increase in cAMP-activated currents upon DHT treatment (Figure S3). No whole Cl− currents could be activated in cells incubated with estrogen.
5. Testosterone Enhances Intracellular Ca2+ Signals
DHT increased the expression of TMEM16A and enhanced ATP-activated whole cell currents, which may be due to enhanced intracellular Ca2+ signaling. We measured the intracellular Ca2+ concentrations in control mCCDcl1 cells, and in cells treated with DHT. Cells were stimulated by different concentrations of ATP, which caused a concentration-dependent increase in intracellular Ca2+ concentration (Figure 6G). Notably, DHT did not further enhance transient Ca2+ increase induced by ATP, but shifted the basal Ca2+ concentration to higher values. This suggests a Ca2+ leakage out of the endoplasmic reticulum (ER) Ca2+ store, or enhanced Ca2+ influx through plasma membrane Ca2+ channels (Figure 6G).
We applied a Ca2+ store release protocol to empty the ER Ca2+ store by inhibiting the sarcoplasmic endoplasmic reticulum ATPase (SERCA), using cyclopiazonic acid (CPA). Remarkably, removal of extracellular Ca2+ lead to a more pronounced decrease in basal cytosolic Ca2+ in DHT, but not in EST-treated cells (Figure 7A, left and middle panel). Subsequent addition of CPA induced a more pronounced Ca2+ store release in DHT-treated cells, and a largely augmented Ca2+ influx upon re-addition of extracellular Ca2+ (Figure 7A, left and right panel). Thus, Ca2+ store release and store operated Ca2+ influx were augmented in DHT-treated cells. We analyzed the expression of a number of Ca2+ transporting proteins such as inositol trisphosphate receptors (IP3R1-3), the Ca2+ influx channels Orai1, TRPC1, TRPV4, and the ER Ca2+ sensor stromal interaction molecule 1 (Stim1). Notably, expression of a number of Ca2+ channels and Stim1 were augmented by both DHT and EST incubated cells. As shown in Figure 6C, DHT enhanced the expression of TMEM16A, while EST had no significant effect on TMEM16A-expression (data not shown). By comparing Ca2+ signals obtained from male and female primary renal epithelial cells, we also found higher basal Ca2+ levels in male cells, along with larger ATP-induced Ca2+ transients. Taken together, the more severe cystic phenotype found in males is likely to be caused by enhanced cell proliferation possibly due to enhanced basal intracellular Ca2+ levels, which is probably due to enhanced expression of Ca2+ transporting/regulating proteins.
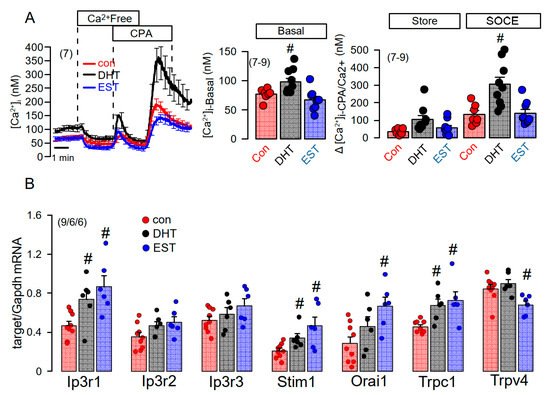
Figure 7. Effect of dihydrotestosterone and estrogen on intracellular Ca2+ concentrations and expression of Ca2+-regulating proteins in mouse mCCDcl1 cortical collecting duct cells. (A) Summary time course of the effects of extracellular Ca2+-free buffer and cyclopiazonic acid (10 µM) on intracellular Ca2+ concentrations in control mCCDcl1 cells and mCCDcl1 cells treated with dihydrotestosterone (DHT; 10 µM, 24 h) and estrogen (EST, 10 µM; 24 h). Summary of basal intracellular Ca2+ concentration and changes in intracellular Ca2+ induced by CPA (Ca2+ store release through leakage channels; Store) and re-addition of Ca2+ to the extracellular buffer (store operated Ca2+ entry; SOCE). (B) RT-PCR analysis of the expression of Ca2+-regulating proteins and effects of DHT and EST. Mean ± SEM (number of experiments in each series). # Significant increase when compared with con (p < 0.05; ANOVA).
References
- Gabow, P.A.; Johnson, A.M.; Kaehny, W.D.; Kimberling, W.J.; Lezotte, D.C.; Duley, I.T.; Jones, R.H. Factors affecting the progression of renal disease in autosomal-dominant polycystic kidney disease. Kidney Int. 1992, 41, 1311–1319.
- Stewart, J.H. End-stage renal failure appears earlier in men than in women with polycystic kidney disease. Am. J. Kidney Dis. 1994, 24, 181–183.
- Johnson, A.M.; Gabow, P.A. Identification of patients with autosomal dominant polycystic kidney disease at highest risk for end-stage renal disease. J. Am. Soc. Nephrol. 1997, 8, 1560–1567.
- Cowley, B.D., Jr.; Rupp, J.C.; Muessel, M.J.; Gattone, V.H., 2nd. Gender and the effect of gonadal hormones on the progression of inherited polycystic kidney disease in rats. Am. J. Kidney Dis. 1997, 29, 265–272.
- Dubey, R.K.; Jackson, E.K. Estrogen-induced cardiorenal protection: Potential cellular, biochemical, and molecular mechanisms. Am. J. Physiol. Ren. Physiol. 2001, 280, F365–F388.
- Stringer, K.D.; Komers, R.; Osman, S.A.; Oyama, T.T.; Lindsley, J.N.; Anderson, S. Gender hormones and the progression of experimental polycystic kidney disease. Kidney Int. 2005, 68, 1729–1739.
- Gretz, N.; Ceccherini, I.; Kränzlin, B.; Klöting, I.; Devoto, M.; Rohmeiss, P.; Hocher, B.; Waldherr, R.; Romeo, G. Gender-dependent disease severity in autosomal polycystic kidney disease of rats. Kidney Int. 1995, 48, 496–500.
- Grantham, J.J.; Ye, M.; Gattone, V.H., 2nd; Sullivan, L.P. In vitro fluid secretion by epithelium from polycystic kidneys. J Clin. Invest. 1995, 95, 195–202.
- Hanaoka, K.; Devuyst, O.; Schwiebert, E.M.; Wilson, P.D.; Guggino, W.B. A role for CFTR in human autosomal dominant polycystic kidney disease. Am. J. Physiol. 1996, 270, C389–C399.
- Davidow, C.J.; Maser, R.L.; Rome, L.A.; Calvet, J.P.; Grantham, J.J. The cystic fibrosis transmembrane conductance regulator mediates transepithelial fluid secretion by human autosomal dominant polycystic kidney disease epithelium in vitro. Kidney Int. 1996, 50, 208–218.
- Buchholz, B.; Teschemacher, B.; Schley, G.; Schillers, H.; Eckardt, K.U. Formation of cysts by principal-like MDCK cells depends on the synergy of cAMP- and ATP-mediated fluid secretion. J. Mol. Med. 2011, 89, 251–261.
- Yang, B.; Sonawane, N.D.; Zhao, D.; Somlo, S.; Verkman, A.S. Small-Molecule CFTR Inhibitors Slow Cyst Growth in Polycystic Kidney Disease. J. Am. Soc. Nephrol. 2008, 19, 1300–1310.
- Magenheimer, B.S.; St John, P.L.; Isom, K.S.; Abrahamson, D.R.; De Lisle, R.C.; Wallace, D.P.; Maser, R.L.; Grantham, J.J.; Calvet, J.P. Early embryonic renal tubules of wild-type and polycystic kidney disease kidneys respond to cAMP stimulation with cystic fibrosis transmembrane conductance regulator/Na(+),K(+),2Cl(-) Co-transporter-dependent cystic dilation. J. Am. Soc. Nephrol. 2006, 17, 3424–3437.
- Xu, N.; Glockner, J.F.; Rossetti, S.; Babovich-Vuksanovic, D.; Harris, P.C.; Torres, V.E. Autosomal dominant polycystic kidney disease coexisting with cystic fibrosis. J. Nephrol. 2006, 19, 529–534.
- O’Sullivan, D.A.; Torres, V.E.; Gabow, P.A.; Thibodeau, S.N.; King, B.F.; Bergstralh, E.J. Cystic fibrosis and the phenotypic expression of autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 1998, 32, 976–983.
- Persu, A.; Devuyst, O.; Lannoy, N.; Materne, R.; Brosnahan, G.; Gabow, P.A.; Pirson, Y.; Verellen-Dumoulin, C. CF gene and cystic fibrosis transmembrane conductance regulator expression in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2000, 11, 2285–2296.
- Wilson, P.D.; Hovater, J.S.; Casey, C.C.; Fortenberry, J.A.; Schwiebert, E.M. ATP release mechanisms in primary cultures of epithelia derived from the cysts of polycystic kidneys. J. Am. Soc. Nephrol. 1999, 10, 218–229.
- Schwiebert, E.M.; Wallace, D.P.; Braunstein, G.M.; King, S.R.; Peti-Peterdi, J.; Hanaoka, K.; Guggino, W.B.; Guay-Woodford, L.M.; Bell, P.D.; Sullivan, L.P.; et al. Autocrine extracellular purinergic signaling in epithelial cells derived from polycystic kidneys. Am. J. Physiol. Ren. Physiol. 2002, 282, F763–F775.
- Kraus, A.; Grampp, S.; Goppelt-Struebe, M.; Schreiber, R.; Kunzelmann, K.; Peters, D.J.; Leipziger, J.; Schley, G.; Schodel, J.; Eckardt, K.U.; et al. P2Y2R is a direct target of HIF-1alpha and mediates secretion-dependent cyst growth of renal cyst-forming epithelial cells. Purinergic Signal. 2016, 12, 687–695.
- Buchholz, B.; Faria, D.; Schley, G.; Schreiber, R.; Eckardt, K.U.; Kunzelmann, K. Anoctamin 1 induces calcium-activated chloride secretion and tissue proliferation in polycystic kidney disease. Kidney Int. 2014, 85, 1058–1067.
- Buchholz, B.; Schley, G.; Faria, D.; Kroening, S.; Willam, C.; Schreiber, R.; Klanke, B.; Burzlaff, N.; Kunzelmann, K.; Eckardt, K.U. Hypoxia-Inducible Factor-1a Causes Renal Cyst Expansion through Calcium-Activated Chloride Secretion. J. Am. Soc. Nephrol. 2014, 25, 465–474.
- Schreiber, R.; Buchholz, B.; Kraus, A.; Schley, G.; Scholz, J.; Ousingsawat, J.; Kunzelmann, K. Lipid peroxidation drives renal cyst growth in vitro through activation of TMEM16A. J. Am. Soc. Nephrol. 2019, 30, 228–242.
- Cabrita, I.; Buchholz, B.; Schreiber, R.; Kunzelmann, K. TMEM16A drives renal cyst growth by augmenting Ca(2+) signaling in M1 cells. J. Mol. Med. 2020, 98, 659–671.
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A in vivo. Nat. Commun. 2020, 11, 4320.
- Lantinga-van Leeuwen, I.S.; Leonhard, W.N.; van de Wal, A.; Breuning, M.H.; Verbeek, S.; de Heer, E.; Peters, D.J. Transgenic mice expressing tamoxifen-inducible Cre for somatic gene modification in renal epithelial cells. Genesis 2006, 44, 225–232.
- Cabrita, I.; Talbi, K.; Kunzelmann, K.; Schreiber, R. Loss of PKD1 and PKD2 share common effects on intracellular Ca2+ signaling. Cell Calcium 2021, 97, 102413.
- Nagao, S.; Kusaka, M.; Nishii, K.; Marunouchi, T.; Kurahashi, H.; Takahashi, H.; Grantham, J. Androgen receptor pathway in rats with autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 2005, 16, 2052–2062.
- Gaeggeler, H.P.; Gonzalez-Rodriguez, E.; Jaeger, N.F.; Loffing-Cueni, D.; Norregaard, R.; Loffing, J.; Horisberger, J.D.; Rossier, B.C. Mineralocorticoid versus glucocorticoid receptor occupancy mediating aldosterone-stimulated sodium transport in a novel renal cell line. J. Am. Soc. Nephrol. 2005, 16, 878–891.
More
Information
Subjects:
Pathology
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
1.2K
Revisions:
2 times
(View History)
Update Date:
22 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No