Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Cory J. Xian | + 2094 word(s) | 2094 | 2021-07-22 10:19:58 | | | |
| 2 | Camila Xu | Meta information modification | 2094 | 2021-08-12 05:05:50 | | | | |
| 3 | Yali Zhang | Meta information modification | 2094 | 2021-08-12 10:59:48 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Xian, C.; Zhang, Y. BMSC Osteogenic or Adipogenic Differentiation. Encyclopedia. Available online: https://encyclopedia.pub/entry/13079 (accessed on 08 February 2026).
Xian C, Zhang Y. BMSC Osteogenic or Adipogenic Differentiation. Encyclopedia. Available at: https://encyclopedia.pub/entry/13079. Accessed February 08, 2026.
Xian, Cory, Yali Zhang. "BMSC Osteogenic or Adipogenic Differentiation" Encyclopedia, https://encyclopedia.pub/entry/13079 (accessed February 08, 2026).
Xian, C., & Zhang, Y. (2021, August 12). BMSC Osteogenic or Adipogenic Differentiation. In Encyclopedia. https://encyclopedia.pub/entry/13079
Xian, Cory and Yali Zhang. "BMSC Osteogenic or Adipogenic Differentiation." Encyclopedia. Web. 12 August, 2021.
Copy Citation
Bone marrow stromal cells (BMSCs) are multipotent cells in the bone marrow which can differentiate into chondrocytes, osteoblasts, adipocytes (fat cells) and other cell types. Many factors have been identified to control the process of BMSC osteogenic or adipogenic differentiation in the bone.
BMSC differentiation
bone/fat formation
microRNAs
1. Introduction
Bone marrow mesenchymal stem cells (BMSCs) have the potential and capacity to differentiate into multiple cell types, such as chondrocytes, osteoblasts, and adipocytes [1][2]. The commitment and differentiation of BMSCs are tightly controlled by factors of their micro-environment, as changed signals and molecules can lead to their abnormal differentiation causing changes in volumes of trabecular bone and bone marrow fat. Bone loss, which is replaced with increased bone marrow fat, occurs under pathological conditions, including aging, menopause, chronic glucocorticoid treatment, or cancer chemotherapy treatment [3][4]. This phenomenon has been shown to occur due to a shift in BMSC differentiation to favour the formation of fat cells or adipocytes (adipogenesis) over bone-forming cells or osteoblasts (osteogenesis). Under these conditions, there are some similarities in the pathological stresses characterized by suppressed osteogenic signals and elevated adipogenic signals, leading to reduced osteogenesis and enhanced adipogenesis differentiation. This change is associated with the development of osteoporosis and increased fracture risk [5][6]. While numerous studies have described the cellular mechanisms of bone/fat balance in the bone marrow [4][7][8], the underlying mechanisms for unbalanced bone/fat formation from the BMSCs has not been fully established.
Osteogenesis or adipogenesis arises due to BMSC differentiation responding to activation of specific signalling pathways. Many recent or current investigations have been trying to identify key factors that can control the activation or attenuation of major signalling pathways and transcription factors in BMSCs. MicroRNAs are 19–25 non-coding RNA molecules that play essential functions in RNA silencing and post-transcriptional regulation of encoding genes by interacting with the 3’ untranslated region (3’ UTR) of the mRNA transcripts that present similar amino acid sequences to inhibit or degrade the target genes [9][10]. MicroRNAs are critical mediators of many biological processes, including tissue formation, cell differentiation, and cancer development [11][12]. Currently, knowledge is continuously accumulating related to microRNA involvement in BMSC differentiation regulation [13][14][15]. By conducting microRNA array and profiling/pathway analyses, certain microRNAs and their specific targets have been identified to control the process of BMSC differentiation, which has led to the elucidation of the functions and regulations of some microRNAs in regulating bone/fat formation [16][17].
2. Key Signalling Pathways Involved in Bone/Fat Formation
Two key signalling pathways have so far been identified to control bone or fat formation: namely, the Wnt/β-catenin and the TGF-β/BMP/Smad signalling pathways.
2.1. Wnt/β-Catenin Signalling
The first observation indicating the link between bone biology and Wingless (Wnt)/β-catenin signalling was made more than ten years ago [18]. Yavropoulou and Yovos studied Wnt signalling mutants and found that these mutants lead to excessive bone formation [19]. Additionally, Wnt/β-catenin has been identified as an essential regulatory signalling pathway for adipogenesis [20][21][22]. Activated Wnt/β-catenin signalling stimulates osteogenic gene expression, including runt-related transcription factor 2 (RUNX2), distal-less homeobox 5 (DLX5), and osterix (OSX) [23][24]. However, inhibition of Wnt/β-catenin signalling induces adipogenic differentiation via increasing the expression of adipogenic-related genes, such as peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT/enhancer-binding protein α (C/EBPα) [25][26]. Further studies have indicated that β-catenin interacts with PPARγ resulting in β-catenin degradation, which attenuates Wnt/β-catenin signalling and bone formation [27]. Therefore, Wnt/β-catenin signalling is well-characterized and strongly implicated in bone/fat regulation (Figure 1).
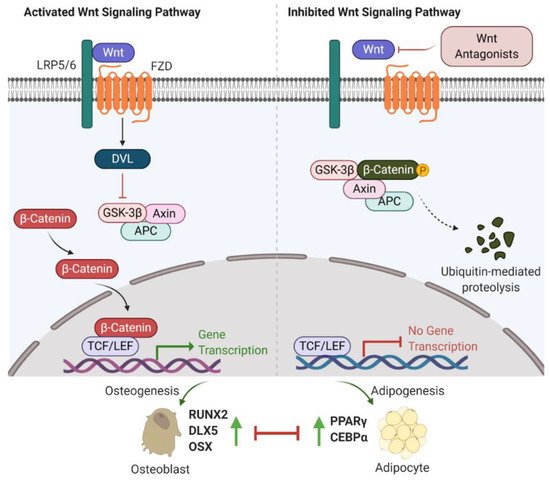
Figure 1. Wnt/β-catenin signalling involved in bone or fat regulation. (Left) In case Wnt ligand is binding to co-receptor complex, β-catenin is released from the destruction complex composed of GSK-3β, Axin and APC. This allows β-catenin to be accumulated and translocated into nucleus. By interacting with TCF/LEF transcription factors, β-catenin activates the osteogenic-related gene transcription program and promotes osteogenesis and bone formation. (Right) In case Wnt ligand is not able to bind to its co-receptor complex (e.g., in the presence of Wnt antagonists (sFRP-1, WIF-1, DKKs and sclerostin)), β-catenin is sequestered by the degradation complex, phosphorylated and subsequently degraded. Attenuated β-catenin enhances adipogenesis and marrow fat formation. Wnt: Wingless; LRP5/6: low-density lipoprotein receptor-related protein 5/6; Frizzled: FZD; DVL: disheveled; GSK-3β: glycogen synthase kinase 3β; APC: adenomatous polyposis coli; TCF/LEF: T-cell factor/lymphoid enhancer-binding factor; RUNX2: runt-related transcription factor 2; DLX5: distal-less homeobox 5; OSX: osterix; PPARγ: peroxisome proliferator-activated receptor γ; C/EBPα: CCAAT/enhancer-binding protein α.
Recent investigations into Wnt/β-catenin signalling have found that at least 19 Wnt ligands (e.g., Wnt1, 2, 3a, 3b, 4, 8, and 10b) can activate this pathway [28]. Activation of Wnt/β-catenin signalling starts with the Wnt ligands binding to a receptor complex that is composed of specific frizzled (FZD) seven-transmembrane-span protein and low-density lipoprotein receptor-related protein 5/6 (LRP-5/6) [12]. Intracellular disheveled (DVL) is then activated, disrupting the inhibitory effects on β-catenin, leading to translocation of β-catenin into the nucleus [29][30]. Intranuclear β-catenin then forms a transcriptional complex with the members of T-cell factor (TCF)/lymphoid enhancer-binding factor (LEF) to together mediate target gene expression [29]. In the absence of Wnt ligands, β-catenin is inhibited by a degradation complex, including glycogen synthase kinase 3β (GSK-3β), axin, and adenomatous polyposis coli (APC) [31]. In addition, the Wnt signalling pathway is negatively regulated by secreted Wnt antagonists. Secreted frizzled-related protein 1 (sFRP-1) and Wnt inhibitory factor 1 (WIF-1) inhibit the interactions between Wnts and the FZD receptor [32]. Sclerostin and Dickkopf (Dkk) families, including Dkk-1, -2, and -4, bind to LRP-5/6, inhibiting LRP-5/6 co-receptor activity [33].
2.2. TGF-β/BMP/Smad Signalling
Transforming growth factor β (TGF-β)/Smad signalling favours early differentiation of osteoprogenitors by promoting DLX5 expression [34][35][36]. DLX5 then induces the expression of RUNX2 and OSX [37]. However, TGF-β/Smad signalling is a double-edged sword in bone homeostasis. Active TGF-β/Smad signalling negatively contributes to osteoblast maturation, mineralization, and osteocyte transition [36].
Bone morphogenetic proteins (BMPs), as members of TGF-β superfamily, also have a strong modulatory effect on osteogenesis/adipogenesis differentiation. Based on the homology of amino acid sequences, bone-inducing BMPs have been divided into four subgroups: BMP2/4, BMP5/6/7, BMP9/10, and BMP12/13/14 subgroups [38]. Other BMPs have not shown osteogenic features but act differentially. For example, BMP3 is an antagonist of BMP2/4 signalling [39]. While BMP/Smad signalling enhances each step of osteogenesis and differentiation into osteocytes [36], BMP/Smad signalling is also known to promote adipogenesis by increasing the expression of PPARγ, a master transcription factor of fat formation [40]. Unlike BMP/Smad signalling, TGF-β/Smad signalling has shown inhibitory effects on adipogenesis and marrow fat formation [36]. Therefore, TGF-β or BMP signalling has multiple roles in regulating bone/fat homeostasis (Figure 2).
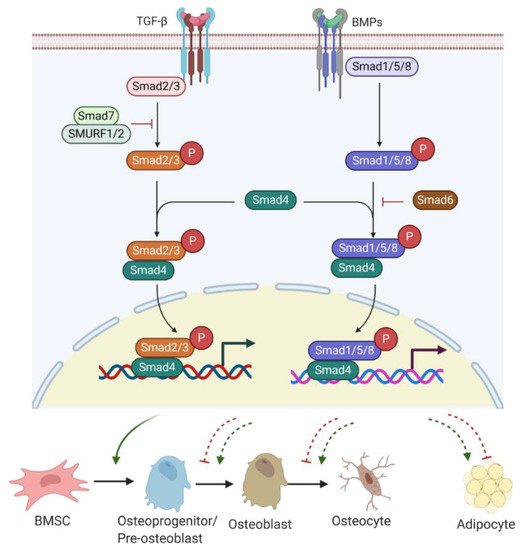
Figure 2. TGF-β/BMP signalling involved in bone or fat regulation. Active TGF-β or BMP binds to TGF-βR I/II or BMPR I/II receptor complex and can induce Smad-dependent signalling. R-Smads (Smad2/3 for TGF-β signalling and Smad1/5/8 for BMPs signalling) complexes with C-Smad, Smad4 and together translocate into the nucleus, where they regulate target gene expression. Smad7 with SMURF 1/2 negatively regulates Smad-dependent signalling by preventing Smad2/3 phosphorylation. Also, Smad6 inhibits the R-Smads/Smad4 complex to disrupt BMP signalling. TGF-β/Smad signalling promotes early differentiation of osteoprogenitors while it represses osteoblast maturation, mineralization, and transition into osteocytes. BMP-Smad signalling promotes almost every step during osteoblast differentiation and induces PPARγ expression for adipogenesis. However, TGF-β/Smad signalling negatively regulates adipogenesis and marrow fat formation. TGF-β: transforming growth factor β; BMPs: bone morphogenetic proteins; TGF-βR: TGF-β receptor; BMPR: BMP receptor; R-Smads; receptor-Smads; C-Smads: common-Smads; I-Smads: inhibitory-Smads; SMURF 1/2: Smad ubiquitin regulatory factor 1 and 2.
TGF-β/BMP ligands, once bound to TGF-β receptor (TGF-βR) I/II or BMP receptor (BMPR) I/II complex, can trigger Smad-dependent signalling pathways. Three categories of Smads are involved in TGF-β/BMPs signal transduction, containing receptor-Smads (R-Smads), common-Smads (C-Smads), and inhibitory-Smads (I-Smads) [41]. R-Smads, including Smads 1, 5, and 8 for the BMPs signalling pathway and Smads2 and 3 for the TGF-β signalling pathway, are activated through phosphorylation. Upon activation, R-Smads release from the receptor complex and form a complex with C-Smad called Smad4 [42]. The complex R-Smads/C-Smad is translocated into the nucleus and modulates the gene expression of diverse transcriptional factors. I-Smads negatively regulate the TGF-β/BMPs signalling pathway. For example, Smad6 can competitively bind to R-Smads, preventing the R-Smads/C-Smad complex formation [43]. Smad7 interacts with E3 ubiquitin ligase proteins, such as Smad ubiquitin regulatory factor 1 and 2 (SMURF 1/2), resulting in degradation of R-Smads [43].
3. Key Factors and Markers Involved in Bone or Fat Regulation
3.1. Key Factors and Markers Involved in Osteogenesis and Bone Formation
Bone formation is a highly restricted process associated with the commitment of numerous BMSCs towards osteoprogenitor cells, followed by the formation of pre-osteoblasts and mature osteoblasts. Many studies have been conducted to understand the factors regulating gene expression during BMSC commitment, osteoblast differentiation, and maturation.
BMPs, members of TGF-β and osteoblast-specific transcription factor OSX are considered important factors to control the commitment of MSCs to the osteoblast lineage [44]. RUNX2 is required for the proliferation of osteoprogenitors and is considered the earliest transcription factor to determine the osteoblast lineage [45][46]. Once osteoprogenitor cells begin to differentiate into pre-osteoblasts, there is a proliferation phase, marked by the high expression of alkaline phosphatase (ALP), a vital enzyme to initiate and regulate bone mineralization [47]. The expression level of ALP is upregulated depending on osteoblast maturation, and thus ALP is considered as a specific molecular marker for the early stage of osteoblast differentiation [48]. In addition, other transcription factors, such as activating transcription factor 4 (ATF4) and DLX5, are all involved in osteoblast differentiation.
The transition of pre-osteoblasts to mature osteoblasts is marked by the secretion of molecules such as osteocalcin (OCN), bone sialoprotein (BSP), osteopontin (OPN), and type 1 collagen, which is essential for osteoid formation and mineralization [49]. OCN is thought to be related to mature osteoblast activity and, therefore, is seen as a marker for late-stage osteoblastogenesis [50]. Type 1 collagen forms osteoid, which is the matrix upon which osteoblasts stimulate the precipitation of calcium salts and phosphorous within the newly formed osteoid to form bone mineral, or hydroxyapatite [51].
3.2. Key Factors and Markers Involved in Adipogenesis and Fat Formation
Adipocyte differentiation is characterized by changes of expression of a series of specific genes and factors that determine the adipocyte phenotype. These changes in gene expression primarily happen at transcriptional levels, which are reflected at different stages of adipogenesis. Numerous studies have explored the molecular mediators that drive the transition from BMSCs to mature adipocytes. The development of adipocytes can be briefly described into two phases [52][53]. The first phase is the commitment of BMSCs to pre-adipocytes, a process influenced by various extracellular factors, including BMPs, insulin/insulin-like growth factor-1 (IGF-1) and TGF-β [54]. The presence of TGF-β negatively regulates adipogenesis and suppression of adipogenesis might be obtained through TGF-β supplementation [54]. While the morphological difference between precursor cells and pre-adipocytes cannot be identified, pre-adipocytes lose the differentiation potentials into other cell types [54]. The second is known as terminal differentiation, in which the pre-adipocytes obtain the characteristics of mature adipocytes and gradually obtain their physiological functions, such as lipid deposition and transport and secretion of adipocytic factors [55].
Two transcription factors, C/EBPs and PPARγ, are recognized to be involved in pre-adipocyte growth arrest and the promotion of terminal adipocyte differentiation and accumulation of lipid droplets [56][57][58]. Some family members of C/EBP, including C/EBPα, C/EBPβ, C/EBPγ, and C/EBPδ, have been recognized to be expressed in adipocytes. The early induction of C/EBPβ and C/EBPδ stimulates the induction of C/EBPα and PPARγ [52][59]. Activated C/EBPα and PPARγ work cooperatively in adipogenesis by activating other adipogenic-specific genes and lipogenic enzymes. C/EBPγ likely inhibits adipocyte differentiation by inactivation of, and heterodimerization with, C/EBPβ [52][60]. Furthermore, activated PPARγ can promote pre-adipocytes to exit from the cell cycle, triggering increased energy delivery to cells [56][61]. C/EBPα and PPARγ are well characterized as primary transcription factors in adipogenesis and fat formation. Other factors are involved in regulating adipogenesis by modulating C/EBPα and PPARγ expression. Sirtuin I (SIRTI), a histone deacetylase that affects energy homeostasis, inhibits adipogenesis by binding to PPARγ. In SIRTI adipocyte-specific knockout mice, PPARγ was found to be hyperacetylated, which lead to a decrease in PPARγ phosphorylation and increased insulin sensitivity [62].
4. MicroRNAs Involved in Bone/Fat Formation
MicroRNAs are small non-coding RNA molecules that regulate the post-transcriptional gene expression by inhibiting target mRNA translations or promoting transcript degradation [9][10]. MicroRNAs that have been found involved in bone and fat formation regulation are summarized in Figure 3. They function through modulating the two key signalling pathways (the Wnt/β-catenin and the TGF-β/BMP/Smad signalling pathways) and the associated transcription factors (Figure 3).
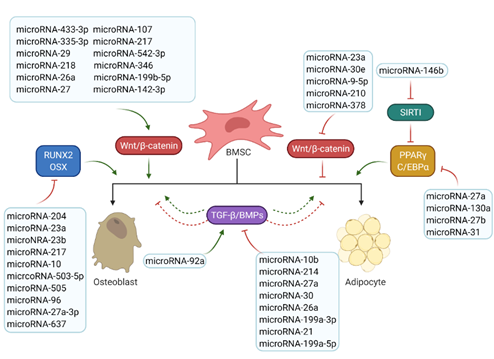
Figure 3. Roles of microRNAs in osteogenesis and adipogenesis. The list of reported microRNAs operates the network of key signalling pathways and factors to regulate bone and marrow fat formation. Green arrows mean positive effects and red arrows represent negative effects.
References
- Bonfield, T.L.; Caplan, A.I. Adult mesenchymal stem cells: An innovative therapeutic for lung diseases. Discov. Med. 2010, 9, 337–345.
- Cook, D.; Genever, P. Regulation of mesenchymal stem cell differentiation. Adv. Exp. Med. Biol. 2013, 786, 213–229.
- Georgiou, K.R.; Scherer, M.A.; Fan, C.-M.; Cool, J.C.; King, T.J.; Foster, B.K.; Xian, C.J. Methotrexate chemotherapy reduces osteogenesis but increases adipogenic potential in the bone marrow. J. Cell. Physiol. 2012, 227, 909–918.
- Li, J.; Zuo, B.; Zhang, L.; Dai, L.; Zhang, X. Osteoblast versus Adipocyte: Bone Marrow Microenvironment-Guided Epigenetic Control. Case Rep. Orthop. Res. 2018, 1, 2–18.
- Veldhuis-Vlug, A.G.; Rosen, C.J. Clinical implications of bone marrow adiposity. J. Intern. Med. 2018, 283, 121–139.
- Rharass, T.; Lucas, S. Mechanisms in endocrinology: Bone marrow adiposity and bone, a bad romance? Eur. J. Endocrinol. 2018, 179, R165–R182.
- Hamam, D.; Ali, D.; Kassem, M.; Aldahmash, A.; Alajez, N.M. MicroRNAs as regulators of adipogenic differentiation of mesenchymal stem cells. Stem Cells Dev. 2015, 24, 417–425.
- Kokabu, S.; Lowery, J.W.; Jimi, E. Cell Fate and Differentiation of Bone Marrow Mesenchymal Stem Cells. Stem Cells Int. 2016, 2016, 3753581.
- Lai, E.C. Micro RNAs are complementary to 3′ UTR sequence motifs that mediate negative post-transcriptional regulation. Nat. Genet. 2002, 30, 363–364.
- Tornesello, M.L.; Faraonio, R.; Buonaguro, L.; Annunziata, C.; Starita, N.; Cerasuolo, A.; Pezzuto, F.; Tornesello, A.L.; Buonaguro, F.M. The Role of microRNAs, Long Non-coding RNAs, and Circular RNAs in Cervical Cancer. Front. Oncol. 2020, 10, 150.
- Tutar, Y. MiRNA and cancer; computational and experimental approaches. Curr. Pharm. Biotechnol. 2014, 15, 429.
- Sun, M.; Zhou, X.; Chen, L.; Huang, S.; Leung, V.; Wu, N.; Pan, H.; Zhen, W.; Lu, W.; Peng, S. The Regulatory Roles of MicroRNAs in Bone Remodeling and Perspectives as Biomarkers in Osteoporosis. Biomed. Res. Int. 2016, 2016, 1652417.
- Tang, P.; Xiong, Q.; Ge, W.; Zhang, L. The role of microRNAs in osteoclasts and osteoporosis. RNA Biol. 2014, 11, 1355–1363.
- Zhao, X.; Xu, D.; Li, Y.; Zhang, J.; Liu, T.; Ji, Y.; Wang, J.; Zhou, G.; Xie, X. MicroRNAs regulate bone metabolism. J. Bone Miner. Metab. 2014, 32, 221–231.
- Ge, D.W.; Wang, W.W.; Chen, H.T.; Yang, L.; Cao, X.J. Functions of microRNAs in osteoporosis. Eur. Rev. Med. Pharmacol. Sci. 2017, 21, 4784–4789.
- Yavropoulou, M.P.; Anastasilakis, A.D.; Makras, P.; Papatheodorou, A.; Rauner, M.; Hofbauer, L.C.; Tsourdi, E. Serum profile of micrornas linked to bone metabolism during sequential treatment for postmenopausal osteoporosis. J. Clin. Endocrinol. Metab. 2020, 105, e2885–e2894.
- Kocijan, R.; Weigl, M.; Skalicky, S.; Geiger, E.; Ferguson, J.; Leinfellner, G.; Heimel, P.; Pietschmann, P.; Grillari, J.; Redl, H.; et al. MicroRNA levels in bone and blood change during bisphosphonate and teriparatide therapy in an animal model of postmenopausal osteoporosis. bioRxiv 2019, 591990.
- Baron, R.; Kneissel, M. WNT signaling in bone homeostasis and disease: From human mutations to treatments. Nat. Med. 2013, 19, 179–192.
- Yavropoulou, M.P.; Yovos, J.G. The role of the Wnt signaling pathway in osteoblast commitment and differentiation. Hormones 2007, 6, 279.
- Arango, N.A.; Szotek, P.P.; Manganaro, T.F.; Oliva, E.; Donahoe, P.K.; Teixeira, J. Conditional deletion of beta-catenin in the mesenchyme of the developing mouse uterus results in a switch to adipogenesis in the myometrium. Dev. Biol. 2005, 288, 276–283.
- Im, D.U.; Kim, S.C.; Chau, G.C.; Um, S.H. Carbamazepine Enhances Adipogenesis by Inhibiting Wnt/β-catenin Expression. Cells 2019, 8, 1460.
- Chen, T.-X.; Cheng, X.-Y.; Wang, Y.; Yin, W. Toosendanin inhibits adipogenesis by activating Wnt/β-catenin signaling. Sci. Rep. 2018, 8, 4626.
- Kennell, J.A.; MacDougald, O.A. Wnt signaling inhibits adipogenesis through beta-catenin-dependent and -independent mechanisms. J. Biol. Chem. 2005, 280, 24004–24010.
- Kang, S.; Bennett, C.N.; Gerin, I.; Rapp, L.A.; Hankenson, K.D.; Macdougald, O.A. Wnt signaling stimulates osteoblastogenesis of mesenchymal precursors by suppressing CCAAT/enhancer-binding protein alpha and peroxisome proliferator-activated receptor gamma. J. Biol. Chem. 2007, 282, 14515–14524.
- Lecarpentier, Y.; Claes, V.; Vallée, A.; Hébert, J.-L. Interactions between PPAR Gamma and the Canonical Wnt/Beta-Catenin Pathway in Type 2 Diabetes and Colon Cancer. PPAR Res. 2017, 2017, 5879090.
- Kawai, M.; Mushiake, S.; Bessho, K.; Murakami, M.; Namba, N.; Kokubu, C.; Michigami, T.; Ozono, K. Wnt/Lrp/beta-catenin signaling suppresses adipogenesis by inhibiting mutual activation of PPARgamma and C/EBPalpha. Biochem. Biophys. Res. Commun. 2007, 363, 276–282.
- Park, I.S.; Kim, B.; Han, Y.; Yang, H.; Cho, U.; Kim, S.I.; Kim, J.H.; Yoon Park, J.H.; Lee, K.W.; Song, Y.S. Decursin and Decursinol Angelate Suppress Adipogenesis through Activation of β-catenin Signaling Pathway in Human Visceral Adipose-Derived Stem Cells. Nutrients 2019, 12, 13.
- Chen, C.; Zhao, M.; Tian, A.; Zhang, X.; Yao, Z.; Ma, X. Aberrant activation of Wnt/beta-catenin signaling drives proliferation of bone sarcoma cells. Oncotarget 2015, 6, 17570–17583.
- Hay, E.; Faucheu, C.; Suc-Royer, I.; Touitou, R.; Stiot, V.; Vayssiere, B.; Baron, R.; Roman-Roman, S.; Rawadi, G. Interaction between LRP5 and Frat1 mediates the activation of the Wnt canonical pathway. J. Biol. Chem. 2005, 280, 13616–13623.
- Medina, M.; Wandosell, F. Deconstructing GSK-3: The Fine Regulation of Its Activity. Int. J. Alzheimers Dis. 2011, 2011, 479249.
- Kobayashi, Y.; Maeda, K.; Takahashi, N. Roles of Wnt signaling in bone formation and resorption. Jpn. Dent. Sci. Rev. 2008, 44, 76–82.
- Zimmerli, D.; Hausmann, G.; Cantu, C.; Basler, K. Pharmacological interventions in the Wnt pathway: Inhibition of Wnt secretion versus disrupting the protein-protein interfaces of nuclear factors. Br. J. Pharmacol. 2017, 174, 4600–4610.
- Krishnan, V.; Bryant, H.U.; Macdougald, O.A. Regulation of bone mass by Wnt signaling. J. Clin. Investig. 2006, 116, 1202–1209.
- Chen, G.; Deng, C.; Li, Y.-P. TGF-β and BMP signaling in osteoblast differentiation and bone formation. Int. J. Mol. Sci. 2012, 8, 272–288.
- Zhao, M.; Qiao, M.; Harris, S.E.; Chen, D.; Oyajobi, B.O.; Mundy, G.R. The zinc finger transcription factor Gli2 mediates bone morphogenetic protein 2 expression in osteoblasts in response to hedgehog signaling. Mol. Cell Biol. 2006, 26, 6197–6208.
- Wu, M.; Chen, G.; Li, Y.-P. TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 2016, 4, 16009.
- Rahman, M.S.; Akhtar, N.; Jamil, H.M.; Banik, R.S.; Asaduzzaman, S.M. TGF-β/BMP signaling and other molecular events: Regulation of osteoblastogenesis and bone formation. Bone Res. 2015, 3, 15005.
- Dumic-Cule, I.; Peric, M.; Kucko, L.; Grgurevic, L.; Pecina, M.; Vukicevic, S. Bone morphogenetic proteins in fracture repair. Int. Orthop. 2018, 42, 2619–2626.
- Wan, D.C.; Kwan, M.D.; Chang, E.I.Y.; Gurtner, G.C.; Longaker, M.T. Chapter 12–Advances in Basic Science Research. In Plastic Surgery Secrets Plus, 2nd ed.; Weinzweig, J., Ed.; Mosby: Philadelphia, PA, USA, 2010; pp. 72–77.
- Lee, M.-J. Transforming growth factor beta superfamily regulation of adipose tissue biology in obesity. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1160–1171.
- Massague, J.; Seoane, J.; Wotton, D. Smad transcription factors. Genes Dev. 2005, 19, 2783–2810.
- Wrana, J.L. Regulation of Smad activity. Cell 2000, 100, 189–192.
- Beederman, M.; Lamplot, J.D.; Nan, G.; Wang, J.; Liu, X.; Yin, L.; Li, R.; Shui, W.; Zhang, H.; Kim, S.H.; et al. BMP signaling in mesenchymal stem cell differentiation and bone formation. J. Biomed. Sci. Eng. 2013, 6, 32–52.
- Bassi, A.; Gough, J.; Zakikhani, M.; Downes, S. 5–Bone tissue regeneration. In Electrospinning for Tissue Regeneration; Bosworth, L.A., Downes, S., Eds.; Woodhead Publishing: Cambridge, UK, 2011; pp. 93–110.
- Kawane, T.; Qin, X.; Jiang, Q.; Miyazaki, T.; Komori, H.; Yoshida, C.A.; Matsuura-Kawata, V.K.d.S.; Sakane, C.; Matsuo, Y.; Nagai, K.; et al. Runx2 is required for the proliferation of osteoblast progenitors and induces proliferation by regulating Fgfr2 and Fgfr3. Sci. Rep. 2018, 8, 13551.
- Komori, T. Regulation of osteoblast differentiation by Runx2. Adv. Exp. Med. Biol. 2010, 658, 43–49.
- Capulli, M.; Paone, R.; Rucci, N. Osteoblast and osteocyte: Games without frontiers. Arch. Biochem. Biophys. 2014, 561, 3–12.
- Aubin, J. Regulation of osteoblast formation and function. Rev. Endocr. Metab. Disord. 2001, 2, 81–94.
- Zoch, M.L.; Clemens, T.L.; Riddle, R.C. New insights into the biology of osteocalcin. Bone 2016, 82, 42–49.
- Nakashima, K.; de Crombrugghe, B. Transcriptional mechanisms in osteoblast differentiation and bone formation. Trends Genet. 2003, 19, 458–466.
- Stewart, J.P.; Shaughnessy, J.D., Jr. Role of osteoblast suppression in multiple myeloma. J. Cell. Biochem. 2006, 98, 1–13.
- Rosen, E.D.; MacDougald, O.A. Adipocyte differentiation from the inside out. Nat. Rev. Mol. Cell Biol. 2006, 7, 885–896.
- Carnevalli, L.S.; Masuda, K.; Frigerio, F.; Le Bacquer, O.; Um, S.H.; Gandin, V.; Topisirovic, I.; Sonenberg, N.; Thomas, G.; Kozma, S.C. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell 2010, 18, 763–774.
- Zhang, K.; Yang, X.; Zhao, Q.; Li, Z.; Fu, F.; Zhang, H.; Zheng, M.; Zhang, S. Molecular Mechanism of Stem Cell Differentiation into Adipocytes and Adipocyte Differentiation of Malignant Tumor. Stem Cells Int. 2020, 2020, 8892300.
- Tang, Q.Q.; Lane, M.D. Adipogenesis: From Stem Cell to Adipocyte. Annu. Rev. Biochem. 2012, 81, 715–736.
- Niemelä, S.; Miettinen, S.; Sarkanen, J.; Ashammakhi, N. Adipose tissue and adipocyte differentiation: Molecular and cellular aspects and tissue engineering applications. Top. Tissue Eng. 2008, 4, 26.
- Gesta, S.; Tseng, Y.H.; Kahn, C.R. Developmental origin of fat: Tracking obesity to its source. Cell 2007, 131, 242–256.
- Gregoire, F.M.; Smas, C.M.; Sul, H.S. Understanding adipocyte differentiation. Physiol. Rev. 1998, 78, 783–809.
- Mandrup, S.; Lane, M.D. Regulating adipogenesis. J. Biol. Chem. 1997, 272, 5367–5370.
- Darlington, G.J.; Ross, S.E.; MacDougald, O.A. The role of C/EBP genes in adipocyte differentiation. J. Biol. Chem. 1998, 273, 30057–30060.
- Fajas, L.; Fruchart, J.C.; Auwerx, J. Transcriptional control of adipogenesis. Curr. Opin. Cell Biol. 1998, 10, 165–173.
- Mayoral, R.; Osborn, O.; McNelis, J.; Johnson, A.M.; Oh, D.Y.; Izquierdo, C.L.; Chung, H.; Li, P.; Traves, P.G.; Bandyopadhyay, G.; et al. Adipocyte SIRT1 knockout promotes PPARγ activity, adipogenesis and insulin sensitivity in chronic-HFD and obesity. Mol. Metab. 2015, 4, 378–391.
More
Information
Subjects:
Biochemistry & Molecular Biology
Contributors
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
2.1K
Revisions:
3 times
(View History)
Update Date:
12 Aug 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No