 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Robert Mark Jackson | + 3026 word(s) | 3026 | 2020-11-06 07:25:03 | | | |
| 2 | Lily Guo | -79 word(s) | 2947 | 2020-11-09 04:41:30 | | | | |
| 3 | Lily Guo | -79 word(s) | 2947 | 2020-11-09 05:06:03 | | |
Video Upload Options
Growth hormone-releasing hormone (GHRH) is secreted primarily from the hypothalamus, but other tissues, including the lungs, produce it locally. GHRH stimulates the release and secretion of growth hormone (GH) by the pituitary and regulates the production of GH and hepatic insulin-like growth factor-1 (IGF-1). Pituitary-type GHRH-receptors (GHRH-R) are expressed in human lungs, indicating that GHRH or GH could participate in lung development, growth, and repair. The goal of thisentry is to present and critically evaluate new findings regarding growth hormone-releasing hormone (GHRH) and its actions in the settings of lung inflammation, fibrosis, and cancer. The essential, unanswered question we address is whether GHRH, as revealed by synthetic peptide probes that activate or inhibit its receptor, plays key roles in lung pathophysiology that are distinct from its effects on growth and metabolism. It provides background on the physiology of GHRH in the lung, which was elucidated using recently developed GHRH receptor peptide agonists and antagonists as mechanistic probes.
1. Introduction
The goal of this entry is to present and critically evaluate new findings regarding growth hormone-releasing hormone (GHRH) and its actions in the settings of lung inflammation, fibrosis, and cancer. The essential, unanswered question we address is whether GHRH, as revealed by synthetic peptide probes that activate or inhibit its receptor, plays key roles in lung pathophysiology that are distinct from its effects on growth and metabolism. It provides background on the physiology of GHRH in the lung, which was elucidated using recently developed GHRH receptor peptide agonists and antagonists as mechanistic probes. While we have used these peptides to identify effects likely attributable to GHRH, we will clearly distinguish these from the established effects of GHRH itself. Animal and cellular models of pulmonary fibrosis, lung cancer, and sarcoid have been developed and exploited to investigate the effects of GHRH-receptors (GHRH-R) inhibition or activation in lung pathophysiology.
We initially present data regarding the physiologic roles of GHRH (and by implication the effects of growth hormone (GH); then, we describe cellular and animal models used to investigate mechanisms of GHRH actions, including effects on gene expression, mitochondrial respiration, and intracellular signaling. Finally, we anticipate future directions for research and describe possible clinical applications of these approaches using synthetic peptides.
To explore GHRH actions, various peptide agonists and antagonists of the receptor have been synthesized in our laboratories[1][2][3]. For example, a GHRH-R antagonist (MIA-602) with the amino acid sequence PhAc–Ada–Tyr–DArg–Asp–Ala–Ile–5FPhe–Thr–Ala–Har–Tyr(Me)–His–Orn–Val–Leu–Abu–Gln–Leu–Ser–Ala–His–Orn–Leu–Leu–Gln–Asp–Ile–Nle–D–Arg–Ha–NH2 has been synthesized by solid phase methods and purified by HPLC. This antagonist and related GHRH agonists (e.g., MR-409) that bind to GHRH-R have been useful in revealing the physiologic and systemic activities of GHRH distinct from its hormonal effects on the pituitary and the GH/insulin-like growth factor-1 (IGF-1) pathway.
The preclinical evaluation of new GHRH analogs of the “Miami” (MIA) series with increased inhibitory potency is well underway. In the synthesis of these analogs, the following substitutions were conserved: D-Arg2, Har9, Abu15, and Nle27. The replacement of Lys12 and Lys21 with Orn increased resistance to enzymatic degradation. The substitution of Arg at positions 11 and 20 by His were conserved. We incorporated pentafluoro-Phe at position 6, Tyr (Me) at position 10, and ω-amino acids at the N-terminus of some analogs. Evaluation of the activity of such analogs on GH release was done in vitro on rat pituitaries and in vivo in male rats. Receptor binding affinities were measured in vitro by competitive binding analysis. The inhibitory activity of analogs on proliferation in vitro was initially tested in several human cancer cell lines such as endometrial adenocarcinoma, colorectal adenocarcinoma, and prostatic carcinoma. Several cell lines were engrafted into nude mice treated subcutaneously with GHRH antagonists at doses of 1–5 μg/day. Analogs MIA-602, MIA-604, MIA-610, and MIA-690 showed high binding affinities to the receptor, which is significantly greater than GHRH(1-29)NH2 itself. The treatment of tumor cells with 5 μM MIA-602 or MIA-690 decreased proliferation by 40–80%. Thus, GHRH analogs of the MIA series suppress cellular proliferation but have relatively weak endocrine GH inhibitory activity. The suppression of cellular growth likely could be induced by the downregulation of GHRH receptors levels[3].
2. Physiological Functions of GHRH
GHRH is a 44-amino acid peptide secreted primarily by the hypothalamus, but various other tissues including the lungs produce it locally[4]. Data reviewed here illustrate the physiologic role of GHRH per se in the lung, which is independent of effects of GHRH-R agonists or antagonists that we describe later. GHRH stimulates the release and secretion of growth hormone (GH) by the pituitary. It stimulates the production of insulin-like growth factor 1 (IGF-1) through the pituitary GH/hepatic IGF-1 axis. GHRH belongs to a peptide family that includes glucagon, secretin, vasoactive intestinal peptide, and pituitary adenylate cyclase-activating peptide[4][5]. The amino terminal sequence of 29 amino acids retains the full biological activity of GHRH. GHRH binds to its receptor on pituitary somatotrophs and activates the synthesis and secretion of growth hormone (GH). GHRH peptide and GHRH-R are expressed in normal extra-pituitary tissues, including tumors, cancer cell lines, and immune cells. A truncated but functional splice variant (SV1) of GHRH-R is found in tumors, pituitary, and peripheral tissues including the lung, implying a physiological role unrelated to its endocrine function.
The existence of a GH-releasing factor in hypothalamic extracts was shown over fifty years ago. Pancreatic tumors can produce GHRH resulting in acromegaly; this observation was made possible by the isolation and characterization of GHRH[6]. The structure of pancreatic GHRH is identical to the hypothalamic peptide. GH acts directly on receptors in peripheral tissues, but in liver, it stimulates the production of insulin-like growth factor 1 (IGF-1), which is a growth-promoting mitogen for human lung fibroblasts[7]. The maintenance of a neuroendocrine axis comprised of GHRH–GH–IGF-1 is the major endocrine function of GHRH. In pituitary and other cells, the effects of GHRH are mediated by binding to specific membrane receptors that belong to the seven-transmembrane class of G-protein coupled receptors. After the binding of GHRH to the GHRH-R of somatotrophs, second messengers include adenylate cyclase–cAMP–protein kinase A, Ca2+–calmodulin, inositol phosphate–diacylglycerol–protein kinase C, and arachidonic acid pathways that result in GH secretion. GHRH-R expression was originally thought to be restricted to pituitary cells due to the tissue-specific expression of transcription factor Pit-1, which plays an essential role in the differentiation of somatotrophs. Alternately spliced variants of GHRH-R such as SV1 have been detected in tissues including the human lung, and some act independently of ligand binding. Therefore, GHRH may have diverse, downstream, paracrine effects.
The enlargement of visceral organs such as the lungs, heart, and kidneys is a cardinal manifestation of acromegaly. Since increased lung volume measured by pulmonary function testing in patients with acromegaly is not related to hyperinflation or to increased inspiratory muscle strength, it is evident that the excess of GH in acromegaly induces the significant growth of adult human lungs[8]. Ectopic acromegaly is rare, and since the discovery of GHRH, few cases have been reported. Tumors secreting GHRH are typically neuroendocrine, mainly of pancreatic or bronchial origin. Patients who present with acromegaly, whose features include enlargement of the lungs, are those of a somatotropic adenoma. GHRH concentration in plasma is specific for diagnosis at a threshold of 250 to 300 ng/L. Somatostatin analogs therapeutically decrease GH secretion and inhibit GHRH secretion.
3. GHRH in the Lung
The GHRH-R modulates activities of key intracellular signaling pathways involved in lung growth, inflammation, and remodeling, demonstrating that GHRH (or GH itself) could participate in lung development, growth, and repair [9][10]. GHRH-R and a splice variant of the receptor are widely distributed in rat tissues other than the hypothalamus and pituitary. GHRH-R was initially detected in rat lung tissue, using RT-PCR after RNA extraction from a whole lung[11]. The GHRH gene is expressed in many lung cells, including alveolar type 2 cells, club cells, and fibroblasts. Higher levels of gene expression are found in lymphocytes and dendritic cells[12]. GHRH-R has been identified as important in the survival of several lung cancer cell lines. Interestingly, both carcinoid tumors and small cell lung cancers produce and release GHRH into the circulation, confirming its presence in lung neuroendocrine cells[6]. As shown in Figure 1, the GHRH-R protein is clearly expressed in normal and diseased human lungs and in normal mouse lungs.
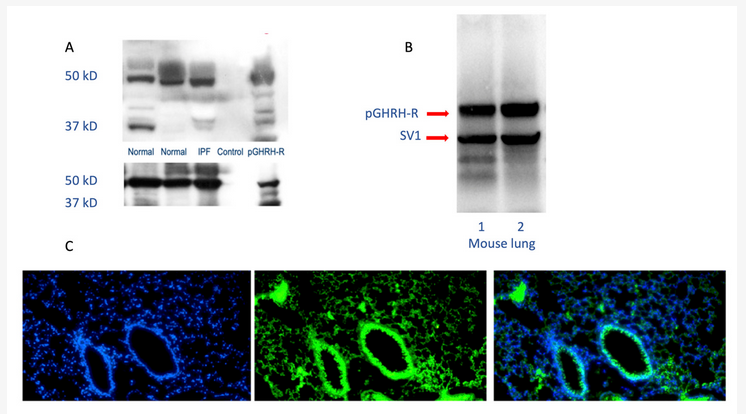
Figure 1. Human and mouse lung westerns (upper panels A and B) and immunofluorescence staining (lower panel C) demonstrate growth hormone-releasing hormone-receptor (GHRH-R) protein. As shown in the upper panels, Western blotting confirms the presence of pituitary-type GHRH-R (pGHRH-R) and splice variant (SV1) in both normal and IPF human lung tissues (upper left panel). Likewise, the pGHRH-R is abundant in lung tissue protein from normal C57BL/6J mice (upper right panel). GHRH-R was detected using a rabbit polyclonal IgG primary antibody (Origene Technologies, Inc., Rockville, MD, USA). As shown in the lower panel, immunofluorescent staining for GHRH-R protein demonstrates prominent expression of the GHRH-R protein in the bronchial epithelium, as well as in alveolar parenchymal cells. (Left, DAPI staining; middle, immunofluorescent antibody to GHRH-R; right, merged images).
Since studies in diverse systems demonstrate GHRH-R in lung cells, and since GHRH-R antagonist modulates inflammation, fibrosis, and lung cancer, GHRH has certain biological importance in the lung and appears to participate in physiologic processes beyond lung injury and repair. Its effects on lung cellular proliferation (e.g., in tumors) are strongly implied by the inhibitory activities of GHRH-R antagonist peptides. Through paracrine signaling, lung cells respond to locally produced GHRH with a variety of effects on proliferation, metabolism, and inflammation[13][14].
3.1. Signaling Pathways
Several peptide GHRH-R agonists and antagonists have been developed in the laboratory; we have utilized some of these to identify specific pathways and their effects regarding the actions of GHRH in lung inflammation, fibrosis, and cancer, as we describe in the section below. Many GHRH-sensitive signaling pathways operate in the lung. These link lung inflammation, mitochondrial function, apoptosis, and fibrosis after injury with the effects of GHRH. Pathways relevant to lung inflammation, fibrosis, and cancer and the effects of GHRH-R antagonists are summarized in Table 1
|
GHRH-R Antagonists |
Model System |
Pathways Implicated in Effects |
Potential Effects |
Reference |
|
MZ-5-156 |
Lung cancer |
AMPK Akt/mTOR ¯ GSK3β ¯ |
Anti-proliferative |
[15] |
|
MIA-602 |
Lung endothelial cells |
ERK ¯ JAK2/STAT3 ¯ p53 AMPK |
anti-inflammatory |
[16] |
|
MIA-602 |
Mouse lung and fibroblasts |
ERK Akt ¯ |
anti-inflammatory, anti-fibrotic, pro-apoptotic |
[34] |
For example, A549 lung epithelial cells, derived from a broncho-avleolar cell tumor, express GHRH-R protein. These lung epithelial cells respond to inhibition of the GHRH-R by the activation of 5′ adenosine monophosphate-activated protein kinase (AMPK) and glycogen synthase kinase 3 B (GSK3B) pathways. GHRH-R antagonist also inhibits protein kinase B (Akt) and mammalian target of rapamycin (mTOR) pathways controlling growth in this cell line, facilitating apoptosis[15]. Thus, GHRH-R antagonists regulate the AMPK pathway and growth in lung cells. Pulmonary artery endothelial cells likewise respond to inhibition of the GHRH-R by the downregulation of extracellular signal related kinase (ERK1/2) and Janus kinase-signal transducer and activator of transcription (JAK2/STAT3) pathways, which are implicated in both lung inflammation and apoptosis[17][18].
Specific signaling pathways modulated by GHRH may be targeted to limit inflammation and post-inflammatory fibrosis, as we summarize in Figure 2. For example, the inhibition of p38 MAPK phosphorylation in C57Bl/6 mice decreases renal fibrosis[19]. The constitutive expression of Akt renders lung tissue susceptible to pulmonary fibrosis[20], in part due to its effects on cellular proliferation[16]. Both pathways are modulated by inhibition of the GHRH-R, as described below.
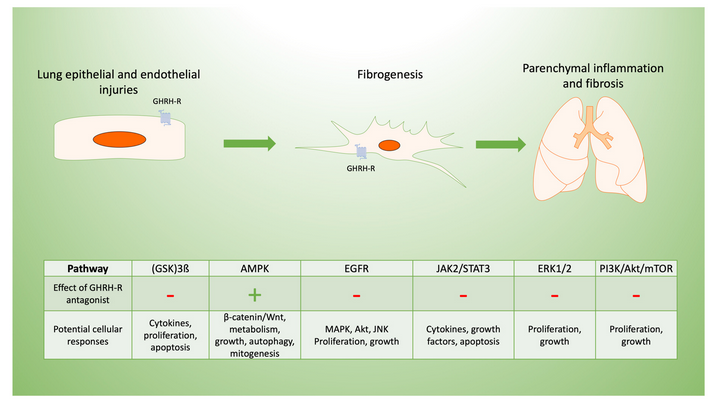
Figure 2. Potential mechanisms of GHRH-R antagonist in lung inflammation and fibrosis. Pulmonary fibrosis typically results from cellular injuries that may be followed by inflammation and progressive fibrosis. GHRH-R is present in lung tissue, and GHRH has effects on lung cellular functions potentially mediated by diverse signaling pathways. These are involved in the lung’s response to inflammation and resulting fibrosis, which may be in response to epithelial injuries as shown diagrammatically above. GHRH-antagonist peptides maintain endothelial barrier function disrupted by inflammation, as they downregulate extracellular signal related kinase (ERK1/2) and Janus kinase-signal transducer and activator of transcription (JAK2/STAT3). In lung epithelial cells, GHRH-R antagonist peptides activate adenosine monophosphate-activated protein kinase (AMPK) and glycogen synthase kinase 3 B (GSK3B) while inhibiting Akt/mammalian target of rapamycin (mTOR) and modulating cellular injury. Similarly, GHRH-R antagonist has anti-proliferative effects in several lung cancer cell lines, which are mediated by epidermal growth factor receptor (EGFR) pathways.
Human fibroblasts express GHRH receptors, which mediate a regulatory effect on proliferation through ERK and Akt signaling. These two signaling cascades are involved in proliferation and apoptosis. Cui and others from our laboratory have reported that when skin wounds in mice are exposed to GHRH agonist, fibroblasts proliferate, and repair of the epithelium is accelerated[1]. Then, fibroblast proliferation induced by a GHRH agonist could be inhibited by blocking ERK. The GHRH receptor is G-protein coupled; ligand binding to GHRH receptor directly leads to cyclic adenosine monophosphate (cAMP)-dependent activation of ERK and Akt pathways. GHRH agonists MR-409 and MR-502 both increase cellular cAMP levels and support cellular growth.
GHRH also stimulates the expression of α-smooth muscle actin (αSMA) through phosphatidylinositol 3-kinase/protein kinase B (PI3K/Akt) signaling, which stimulates contractile activity[21]. ERK activity modulates cellular proliferation by enhancing apoptosis, autophagy, and senescence[18], and GHRH-R antagonist peptides may regulate these activities through downregulation of the ERK pathway.
Microvascular endothelial cells express both the pituitary-type GHRH receptor and SV1. Endothelial cells are responsible for angiogenesis, which is a critical event in wound healing. Uddin and others have shown that GHRH-R peptides suppress the activation of MLC2, ERK1/2, and JAK2/STAT3 and increase p53 and pAMPK, which support the endothelial permeability barrier[22]. GHRH produced by fibroblasts regulates the activities of other cells involved in wound healing in a paracrine fashion. Overall, activation of the GHRH-R provides pro-inflammatory and pro-fibrotic signals, while GHRH-R antagonists modulate inflammation and fibrosis, as described below.
3.2. Fibrogenesis
Transforming growth factor-beta (TGF-β), Wingless and Int-1 (Wnt), hedgehog, Notch, and fibroblast growth factor (FGF) signaling pathways are implicated in regulating lung morphology during development and participate in fibrosis[23]. Wnt/β-catenin signaling is further essential to the regulation of myofibroblast differentiation of mesenchymal cells in the lung and participates in the development of idiopathic pulmonary fibrosis (IPF)[24]. The Wnt/B-catenin pathway is expressed in lung epithelial cells and regulates epithelial and mesenchymal cell interactions during the development of fibrosis. Wnt4 is upregulated by GH and leads to the activation of ERK1 and STAT3, modulating cell growth and survival[25]. Many such pathways are activated during injury and repair and have been linked to post inflammatory fibrosis[26]. Persistent activation, e.g., by TGF-β, can result in lung pathologies, including IPF[27].
The v-AKI murine thymoma viral oncogene/protein kinase B (Akt/PKB) is a serine/threonine-specific kinase involved in apoptosis and proliferation. Akt/PKB regulates cellular survival and metabolism by regulating downstream effectors such as nuclear factor kappa B (NF-κB) and Bcl-2 family proteins in human cancer cells [28][28][27]. Cells respond through Akt/PKB to a variety of cytokines, G protein coupled receptor ligands, and growth factors, making it a potential target to modulate fibrosis. Consistent with this mechanism, the suppression of Akt and mTOR by GHRH-R antagonist treatment inhibits the growth of A549 epithelial cells in culture, and in contrast, GHRH agonists stimulate the Akt pathway.
In addition to its effects on GH and IGF-1, synthetic GHRH-R antagonist MIA-602 inhibits p21-activated kinase 1-signal transducer and activator of transcription 3/nuclear factor-kappa B (PAK1-STAT3/NF-κB), which is consistent with its role in modulating inflammatory and fibrotic processes[29]. GHRH-R antagonist activity is in part modulated by p53 and p21, which suppress NF-KB and inducible nitric oxide synthase (iNOS) while facilitating apoptosis[30][31].
Fibroblast growth factor-2 (FGF-2) is expressed in the epithelium, vascular endothelium, smooth muscle and epithelial basement membrane; and, an increased expression of FGF-2 occurs in lungs from patients with IPF. FGF-receptor regulates fibroblast apoptosis through Akt and focal adhesion kinase (FAK) related to TGF-β activation [13]. Several pro-fibrotic pathways, including Akt and ERK, are inhibited by GHRH-R antagonists including MIA-602, demonstrating that GHRH plays a key role in modulating lung fibrosis following inflammation by blocking these pathways[1][32].
PI3K-Akt signaling inhibits apoptosis and regulates cell growth, survival, and proliferation. Receptor activation recruits PI3K to the inner cell membrane through phosphorylated tyrosine kinases, activated proto-oncogene protein p21 (RAS), or G protein beta and gamma subunits[27]. Akt plays important roles in response to growth factors to regulate metabolism, growth, apoptosis, and survival. In response to injury, sustained PI3K activation worsens lung fibrosis due to bleomycin[33]. The PI3K–Akt pathway is involved in the pathogenesis of fibrosis and regulates epithelial to mesenchymal transition.
MIA-602 has effects on lung fibroblast signal transduction, including anti-apoptotic Akt/protein kinase B (PKB) and pro-growth extracellular signal-regulated kinase (ERK), which are two of the key pathways controlling fibroblast survival and proliferation. Bleomycin increased Akt phosphorylation and concomitantly decreased ERK phosphorylation after an in vitro incubation of lung fibroblasts. In contrast, MIA-602 reduced Akt phosphorylation due to bleomycin and restored ERK activation in lung fibroblasts treated with bleomycin. Thus, inhibition of the GHRH-R appeared to prevent activation of the PI3K–Akt pathway caused by bleomycin, which is consistent with the observed reduction of inflammation and fibrosis[34].
TGF-β and bFGF regulate apoptosis in granulation tissue fibroblasts while inhibiting Akt[27]. The inhibition of GHRH-R by MIA-602 decreases Akt activation after bleomycin and leads to fibroblast apoptosis. GHRH-R antagonist causes apoptosis in epithelial cells through the suppression of mitogen-activated protein kinases (MAPK) and inhibits epithelial to mesenchymal transition, so limiting fibrosis. GHRH inhibitors also act to reduce fibrosis through the suppression of MAPK and p53[31].
References
- Cui, T.; Jimenez, J.; Block, N.; Badiavas, E.; Rodriguez-Menocal, L.; Granda, A.; Schally, A. Agonistic analogs of growth hormone releasing hormone (GHRH) promote wound healing by stimulating the proliferation and survival of human dermal fibroblasts through ERK and Akt pathways. Oncotarget 2016, 7, 52661–52672, https://doi.org/10.18632/oncotarget.11024.
- Schally, A.; Varga, J.; Engel, J. Antagonists of growth-hormone-releasing hormone: An emerging new therapy for cancer. Nat. Rev. Endocrinol 2008, 4, 33–43, https://doi.org/10.1038/ncpendmet06773.
- Zarandi, M.; Cai, R.; Kovacs, M.; Popovics. P.; Szalontay, L.; Cui, T.; Sha, W.; Jaszberenyi, M.; Varga, J.; Zhang, X.; et al. Synthesis and structure-activity studies on novel analogs of human growth hormone releasing hormone (GHRH) with enhanced inhibitory activities on tumor growth. Peptides 2017, 89, 60–70, http://dx.doi.org/10.1016/j.peptides.2017.01.0090196-9781.
- Kiaris, H.; Chatzistamou, I.; Papavassiliou, A.; Schally, A. Growth hormone-releasing hormone: Not only a neurohormone. Trends Endocrinol Metab 22, 311- 317, 2011, https://doi.org/10.1016/j.tem.2011.03.006.
- Rekasi, Z.; Varga, J.; Schally, A.; Halmos, G.; Armatis, P.; Groot, K.; Czompoly, T. Antagonists of growth hormone-releasing hormone and vasoactive intestinal peptide inhibit tumor proliferation by different mechanisms: Evidence from in vitro studies on human prostatic and pancreatic cancers. Endocrinol 2000, 141, 2120–2128, https://doi.org/10.1210/endo.141.6.7511.
- Borson-Chazot, F.; Garby, L.; Raverot, G.; Claustrat, F.; Raverot, V.; Sassolas, G. Acromegaly induced by ectopic secretion of GHRH: A review 30 years after GHRH discovery. Ann. D’endocrinologie 2012, 73, 497–502, https://doi.org/10.1016/j.ando.2012.09.004.
- Warnken, M.; Reitzenstein, U.; Sommer, A.; Fuhrman, M.; Mayer, P.; Enzmann, H.; Juergens, U.; Racke, K. Characterization of proliferative effects of insulin, insulin analogues and insulin-like growth factor-1 in human lung fibroblasts, Arch Pharmacol 2010, 382, 511–525.
- Garcia-Rio, F.; Pino, J.; Diez, J.; Ruiz, A.; Villasante, C.; Villamor, J. Reduction of lung distensibilty in acromegaly after suppression of growth hormone hypersecretion. Am. J. Respir Crit Care Med. 2001, 164, 852–857, https://doi.org/10.1164/ajrccm.164.5.2005059.
- Cao, H.; Wang, C.; Chen, X.; Hou, J.; Xiang, Z.; Shen, Y.; Han, X. Inhibition of Wnt/β-catenin signaling suppresses myofibroblast differentiation of lung resident mesenchymal stem cells and pulmonary fibrosis. Sci Rep. 2018, 8, 13644, doi:10.1038/s41598-018-28968-9.
- Fu, Y.; Arkins, S.; Wang, B.; Kelley, K. A novel role of growth hormone and insulin-like growth factor-1. J. Immunol 1991, 146, 1602–1608, http://www.jimmunol.org/content/146/5/1602.
- Matsubara, S.; Sato, M.; Mizobuchi, M.; Niimi, M.; Takahara, J. Differential gene expression of Growth Hormone (GH)-Releasing Hormone (GRH) and GRH receptor in various rat tissues. Endocrinology 1995, 136, 4147–4150, https://doi.org/10.1210/en.136.9.4147.
- Christidoulou, C.; Schally, A.; Chazistamou, I.; Kondi-Pafiti, A.; Lamnissou, K.; Kouloheri, S.; Kalofoutis, A.; Kiaris, H. Expression of growth hormone-releasing hormone and splice variant of GHRH receptors in normal mouse lung tissues. Regul. Pept. 2006, 136, 105–108, doi:10.1016/j.regpep.2006.05.001.
- Akasaka, Y.; Ono, I.; Kamiya, T.; Ishikawa, Y.; Kinoshita, T.; Ishiguro, S.; Ishii, T. The mechanisms underlying fibroblast apoptosis regulated by growth factors during wound healing. J. Pathol 2010, 22, 285–299, doi:10.1002/path.2710.
- Hung, C.; Rohani, M.; Lee, S.; Chen, P.; Schnapp, L. Role of IGF-1 pathway in lung fibroblast activation. Respir Res. 2013, 14, 102, https://doi.org/10.1186/1465-9921-14-102.
- Siejka, A.; Barabutis, N.; Schally, A. GHRH antagonist MZ-5-156 increases the expression of AMPK in A549 lung cancer cells. Cell Cycle 2011, 10, 3714–3718, doi:10.4161/cc.10.21.17904.
- Zhang, X.; Xing, R.; Chen, L.; Liu, C.; Miao, Z. PI3K/Akt signaling is involved in the pathogenesis of bleomycin-induced pulmonary fibrosis via regulation of epithelial-mesenchymal transition. Molec Med. Rep. 2016, 14, 5699–5706, doi:10.3892/mmr.2016.5960.
- Morrisey, E. Wnt signaling and pulmonary fibrosis. Am. J. Pathol 2003, 162, 1393–1397, doi:10.1016/S0002-9440(10)64271-X.
- Vouyovitch, C.; Perry, J.; Liu, D.; Bezin, L.; Vilain, E.; Diaz, J.; Mertani, H. Wnt4 mediates the autocrine effects of growth hormone in mammary carcinoma cells. Endocr. -Relat. Canc 2016, 23, 571– 585, https://doi.org/10.1530/ERC-15-0528.
- Madtes, D.; Busby, K.; Strandjord, T.; Clark, J. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am. J. Resp Mol. Cell Biol 1994, 11, 540–551, https://doi.org/10.1165/ajrcmb.11.5.7524566.
- Osaki, M.; Oshimura; M., Ito, H. The PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676, doi:10.1023/B:APPT.0000045801.15585.dd.
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2007, 9, 59–71, https://doi.org/10.1111/j.1582-4934.2005.tb00337.x.
- Wei, Y.; Kim, T.; Peng, D.; Duan, D.; Gibbons, D.; Yamauchi, M.; Jackson, J.; Le Saux, C.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. J. Clin. Invest. 2017, 127, 3675–3688, 10.1172/JCI94624.
- Gan, J.; Ke, X., Jiang, J.; Dong, H.; Yao, Z.; Lin, Y.; Lin, W.; Wu, X.; Yan, S.; Zhuang, Y.; et al. Growth hormone-releasing hormone receptor antagonists inhibit human gastric cancer through downregulation of PAK1-STAT3/NF-κB signaling. Proc. Natl Acad Sci Usa 2016, 113, 14745–14750, https://doi.org/10.1073/pnas.1618582114.
- Madtes, D.; Busby, K.; Strandjord, T.; Clark, J. Expression of transforming growth factor-alpha and epidermal growth factor receptor is increased following bleomycin-induced lung injury in rats. Am. J. Resp Mol. Cell Biol 1994, 11, 540–551, https://doi.org/10.1165/ajrcmb.11.5.7524566.
- Wei, Y.; Kim, T.; Peng, D.; Duan, D.; Gibbons, D.; Yamauchi, M.; Jackson, J.; Le Saux, C.; Calhoun, C.; Peters, J.; et al. Fibroblast-specific inhibition of TGF-beta1 signaling attenuates lung and tumor fibrosis. J. Clin. Invest. 2017, 127, 3675–3688, 10.1172/JCI94624.
- Osaki, M.; Oshimura; M., Ito, H. The PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676, doi:10.1023/B:APPT.0000045801.15585.dd.
- Song, G.; Ouyang, G.; Bao, S. The activation of Akt/PKB signaling pathway and cell survival. J. Cell Mol. Med. 2007, 9, 59–71, https://doi.org/10.1111/j.1582-4934.2005.tb00337.x.
- Gan, J.; Ke, X., Jiang, J.; Dong, H.; Yao, Z.; Lin, Y.; Lin, W.; Wu, X.; Yan, S.; Zhuang, Y.; et al. Growth hormone-releasing hormone receptor antagonists inhibit human gastric cancer through downregulation of PAK1-STAT3/NF-κB signaling. Proc. Natl Acad Sci Usa 2016, 113, 14745–14750, https://doi.org/10.1073/pnas.1618582114.
- Barabutis, N.; Schally, A. Antioxidant activity of growth hormone-releasing hormone antagonists in LNCaP human prostate cancer line. Proc. Natl Acad Sci Usa 2008, 105, 20470–20475, doi:10.1073/pnas.0811209106.
- Barabutis, N.; Siejka, A.; Schally, A. Growth hormone releasing hormone induces the expression of nitric oxide synthase. J. Cell. Mol. Med. 2010, 15, 1148–1155, doi:10.1111/j.1582-4934.2010.01096.x.
- Madala, S.; Schmidt, S.; Davidson, C.; Ikegami, M.; Wert, S.; Hardie, W. MEK-ERK pathway modulation ameliorates pulmonary fibrosis associated with epidermal growth factor receptor activation. Am. J. Respir Cell Mol. Biol 2012, 46, 380–388, doi:10.1165/rcmb.2011-0237OC.
- Zhang, C.; Cui, T.; Tian, R.; Cai, R.; Mirsaeidi, M.; Schally, A.; Jackson, R. Growth hormone-releasing hormone receptor antagonist MIA-602 modulates lung inflammation, cellular signal transduction, and promotes apoptosis of mouse lung fibroblasts. European Respiratory Society. https://ers.conference2web.com/- !resources/growth-hormone-releasing-hormone-receptor-antagonist-mia-602-modulates-lung-inflammation-cellular-signal-transduction-and-promotes-apoptosis-of-mouse-lung-fibroblasts (accessed 24 August 2020).
- Königshoff, M.; Balsara, N.; Pfaff, E.; Kramer, M.; Chrobak, I.; Seeger, W.; Eickelberg, O. Functional Wnt signaling is increased in idiopathic pulmonary fibrosis. PLoS ONE 2008, 3, 1–12, doi:10.1371/journal.pone.0002142.
- Zhang, C.; Cai, R.; Lazerson, A.; Delcroix, G.; Wangpaichitr, M.; Mirsaeidi, M.; Griswold, A. J.; Schally, A. V.; Jackson, R. M. Growth hormone-releasing hormone receptor antagonist modulates lung inflammation and fibrosis due to bleomycin. Lung 2019, 197, 541–549, doi:10.1007/s00408-019-00257-w.


