 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Simona Villata | + 4340 word(s) | 4340 | 2020-08-27 10:23:24 | | | |
| 2 | Simona Villata | Meta information modification | 4340 | 2020-09-01 18:18:29 | | | | |
| 3 | Nicole Yin | Meta information modification | 4340 | 2020-09-02 10:57:08 | | |
Video Upload Options
Engineered Extracellular Vesicles are devices obtained through the surface modification of natural extracellular vesicles, both using direct and indirect methods, i.e. engineering of the parental cells.
The aim of their production is to obtain extracellular vesicles that are more reliable in terms of reproducibility and that present some desired features, that can change depending on the application.
Definition of EVs
Every day, in the human body, cells release in the extracellular space particles delimited by a lipid bilayer that cannot replicate. Such particles are defined as extracellular vesicles (EVs)[1] . This general term encompasses a huge number of structures, referred as exosomes, microvesicles, microparticles, ectosomes, oncosomes, apoptotic bodies, and many other names [1], which differ in biogenesis, release pathways, size, content, and function.
1. Introduction
The nomenclature of these vesicles evolved during the last two decades [2]. The widespread and oldest classification divides the EVs on the base of their biogenetic pathway and, even simplistically, identifies three main classes: the exosomes, the microvesicles, and the apoptotic bodies (Figure 1). The exosomes consist of vesicles with an endocytic origin, ranging in size from around 50 to 150 nm. They originate as intraluminal vesicles (ILVs) of the multivesicular bodies (MVBs) and become exosomes when secreted in the extracellular milieu. The microvesicles originate from the direct outwards budding and fission of the plasma membrane and range in size from 50 nm to 1 μm, and in some case they can reach higher dimensions of up to 10 μm (this is the case with the large vesicles released by cancer cells, named oncosomes). Lastly the apoptotic bodies are vesicles resulting from the disassembly of the apoptotic cells, which are generally defined as 500 nm-5 μm in diameter [3].
In recent years, the International Society for Extracellular Vesicles proposed a new classification based on the size range[1]. In fact, as reported by Thery et al.[1], it is extraordinary difficult to assign an EV to a particular biogenesis pathway due to the lack of specific markers; therefore, a classification on a physical characteristic, such as the size, results as being most appropriate. In the most recent publications, EVs are divided into two main classes, defined as small EVs (< 100 nm or < 200 nm) and medium/large EVs (> 200 nm).
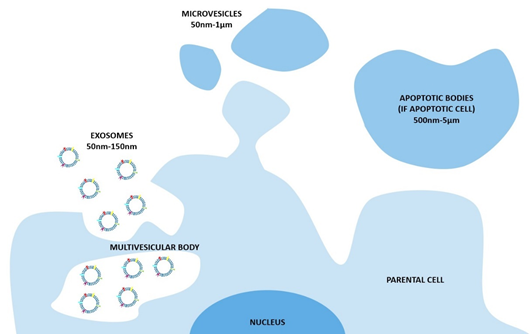
Figure 1. The biogenesis of extracellular vesicles (EVs) and the different pathways according to the current classification. In particular, exosomes consist of vesicles with an endocytic origin, ranging in size from around 50 to 150 nm. They originate as intraluminal vesicles (ILVs) of the multivesicular bodies (MVBs) and become exosomes when secreted in the extracellular milieu. The microvesicles originate from the direct outwards budding and fission of the plasma membrane and range in size from 50 nm to 1 μm. The apoptotic bodies are vesicles resulting from the disassembly of the apoptotic cells, which are generally defined as 500 nm-5 μm in diameter.
After their isolation, EVs can be modified in order to obtain enhanced targeting and biomimetic features[4]. This concept is called engineering of EVs because, starting from naturally-derived EVs, scientists produce a vesicle with the desired behaviour[5]. It is important to highlight that an extracellular vesicle can be modified through both acting on the parental cells (indirect method) and by directly modifying the vesicle once it has been isolated (direct method)[4]. Another important branch of EV engineering is their hybridization after their isolation, where EV membranes are fused with synthetic liposomes[6].
2. Indirect Methods
This method is based on the engineering of parental cells, i.e., the cells that will produce the EVs[6]. First, parental cells can be genetically or metabolically modified to alter the surface expression of the produced EVs and thus enhance their targeting ability and biocompatibility[4]. This can be carried out by inserting the coding sequence of the ligand of interest inframe to the coding sequences between the signal peptide and N-terminus of the mature peptide of a transmembrane protein[5]. Using a retrovirus or a lentivirus as gene transfer vector, this package is transmitted and expressed in parental cells[6]. At this point, these transfected parental cells will produce EVs with the desired peptide expressed on their surface. In Table 1 and Figure 2, some applications of this indirect method are reported [4][6].
Table 1. Applications of membrane functionalization through indirect methods.
|
Parental Cells |
Functionalization |
Cell Engineering Conditions |
Recipient Cells |
Treatment Conditions |
Application |
Reference |
|
HEK293 |
Tetraspanins (CD63, CD9, CD81) |
Transfected at 40~60% confluency using plasmid DNA (1–2 µg/well) for 48h with PureFection Transfection Reagent or FuGENE6 t.r. |
HEK293 |
Cells at confluency of 80% and 50 µg of exosomes |
Tracking, imaging and targeting drug delivery |
[7] |
|
GM-CSF |
Lamp-2b fused to the neuron-specific RVG peptide |
Transfected 4 days using 5 µg of pLamp2b and 5 µl of TransIT LT1 t.r. |
C2C12 and Neuro2A IN VIVO: C57BL/6 mice |
Exosomes (12 µg proteins) and 400 nanomoles of siRNA IN VIVO: i.v. 150 µg of exosomes |
Delivering of siRNA to the brain in mice with a reduced immunogenicity |
[8] |
|
Immaturedendritic cells (imDCs) |
Lamp2b fused to CRGDKGPDC |
Transfected with the vector expressing iRGD-Lamp2b fusion proteins using Lipofectamine 2000 t.r. |
MDA-MB-231 IN VIVO: BALB/c nude mice |
2 mM Dox-loaded exosomes IN VIVO: i.v. EVs 3mg/kg Dox loaded exosomes |
Targeted tumour therapy |
[9] |
|
Neuro2A |
GPI |
Transfected with pLNCX-DAF-R2 or pLNCX-DAF-EGa1 using TransIT 2020 t.r. |
Neuro2A, HeLa, and A431 |
40,000 cells per well or cells at a confluency of 80–90% and EVs at 5 µg/mL |
Promoting tumor cell targeting |
[10] |
|
HEK293 |
GE11 or EGF |
Transfected with pDisplay encoding GE11 or EGF using FuGENE HD t.r. |
HCC70 HCC1954 MCF-7 IN VIVO: RAG2–/– mice |
1 × 105 breast cancer cells and 1 µg of exosomes IN VIVO: i.v. 1 µg of exosomes, once per week for 4 weeks |
Delivering of antitumor microRNA to EGFR-expressing breast cancer cells |
[11] |
|
BT474, SKBR3, HER2+, JAWSII DCs, 4T1-HER2, and bmDCs |
CEA and HER2 coupled to the C1C2 domain of lactadherin |
Transfected with p6mLC1C2 containing either human CEA (nt 1-2025) or human HER2/neu (nt 1-1953) |
IN VIVO: C57BL/6J and BALB/c mice, hCEA or HER2 transgenic mice |
IN VIVO: 2.6 × 1010 or 5.2 × 109 or 1.05 × 109 viral particles |
Increasing vaccine potency |
[12] |
|
HEK293-F, E6, and CT26 |
PSA and PAP coupled to the C1C2 domain of lactadherin |
Transfected with pPSA/Zeo, pPSA-C1C2/Zeo, pPAP/Hygro, or pPAP-C1C2/Hygro using Lipofectamine LTX reagent and PLUS Reagent |
IN VIVO: Male BALB/c or C57BL/6 mice |
IN VIVO: 5E7 TCID50 of the MVA-BN-PRO viral vectors once every 2 weeks for a total of three treatments |
Targeting of tumor antigens to improve antigen immunogenicity and therapeutic efficacy |
[13] |
|
DCs |
C1C2 domain of lactadherin |
Transfected with modified p6mLC1C2 or pcDNA6-Myc/His using Fugene 6 t.r. |
IN VIVO: Balb/C mice
|
IN VIVO: six inoculums of YAC exosomes with HLA-A2 or five inoculums of YAC/HLA-A2 exosomes with pMAGE-A3 |
Usage of antibodies against tumor biomarkers to attach the drug target candidates |
[14] |
|
THP-1 |
RGD- DSPE-PEG and/or DSPE-PEG-SH |
Incubated with DSPE-PEG-SH and/or DSPE-PEG-RGD for 2 days |
MCF-7 and HeLa IN VIVO: tumor-bearing mouse |
4 × 105 cells/mL and 100 µL per well of 50 µg/mL exosomes IN VIVO: i.v. 200 µL of exosomes at 5 mg/mL |
Active targeted chemo‐photothermal synergistic tumor therapy |
[15] |
|
THP-1 |
DSPE-PEG-biotin and/or DSPE-PEG-FA |
Incubated with DSPE-PEG-biotin and/or DSPE-PEG-folate for 2 days |
HeLa IN VIVO: C57BL/6 mice |
40 μg/mL of EVs IN VIVO: i.v. EVs with a total of 1.16 mg iron |
Rapid isolation and enhanced tumor targeting |
[16] |
|
Cal 27 cells |
DSPE-PEG-biotin and DSPE-PEG-folate |
Incubated with DSPE-PEG-biotin and DSPE-PEG-folate |
MDA-MB-231 IN VIVO: BALB/C mice |
Series of dose and concentration IN VIVO: 18–22 g of EVs via the tail vein |
Enhanced target and synergistic therapy for breast cancer |
[17] |
|
HUVECs |
DSPE-PEG-biotin (to then attach SA-QDs) |
Cultured with DSPE-PEG-biotin for several days and then incubated with SA-QDs |
EPCs IN VIVO: nude mice bearing A2058 xenografts |
Short-term incubation IN VIVO: injection |
Antitumor siRNA delivery |
[18] |
|
HUVECs |
DSPE-PEG-biotin and SA-FITC |
Incubated in modified medium containing 40 µg/mL DSPE-PEG-biotin for several days |
HepG2 and 3T3 fibroblast IN VIVO: cervical cancer-bearing male BALB/c mice |
5 × 103 cells per well and 0, 10, 40, 80, 100, and 200 mg/mL of exosomes IN VIVO: exosomes at 5 mg/mL, 200µL per mice |
Active targeted drug delivery to tumor cells |
[19] |
|
HEK 293T cells |
GlucB with sshBirA to conjugate streptavidin–Alexa 680 |
Transduced with lentivirus vectors, CSCW-Gluc-IRES-GFP or CSCW-GlucB-IRES-GFP, then infection with CSCW-sshBirA-IRES-mCherry lentiviruses |
IN VIVO: athymic nude mice spiked with EV-GlucB |
IN VIVO: injected with a bolus of 100 μg EV-GlucB via retro-orbital vein or via tail vein |
Multimodal imaging in vivo, as well as monitoring of EV levels in the organs and biofluids |
[20] |
|
B16BL6 |
Streptavidin–lactadherin and biotinylated GALA |
4 × 106 cells per dish transfected with the plasmid vector pCMVSAV−LA |
MHC class I molecules of DCs |
5 × 104 cells per well and exosomes (1 μg of protein) diluted in 0.1 mL of Opti-MEM |
Efficient cytosolic delivery of exosomal tumor antigens |
[21] |
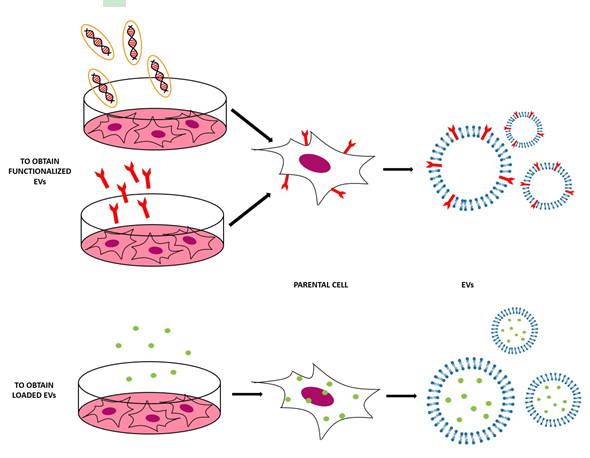
Figure 2. Scheme of the indirect methods used to engineer the EVs, both to functionalize EVs with the molecules of interest (to obtain EVs that expose these molecules on their surface) and to obtain EVs loaded with the desired cargo.
Secondly, parental cells can be incubated with drugs or drug-loaded (or even gene-loaded) nanoparticles (NPs) in a sublethal concentration [4]—after a certain period of time, the therapeutic molecules or NPs will be internalized into the cells and then these cells will produce EVs containing a certain fraction of drug or drug-loaded NPs [4]. In this case, the loading of the cargo is obtained through the engineering of the parental cells[5]. For example, mesenchymal stromal cells (MSCs) can acquire strong anti-tumor activity after priming with paclitaxel (PTX) because MSCs secrete a high amount of membrane microvesicles that will contain the drug[22]. Another study reported how melanoma cells can be loaded with survivin T34A and gemcitabine to produce exosomes that carry the drug to treat pancreatic adenocarcinoma[23]. Doxorubicin and methotrexate have been loaded into tumoral cells and their apoptotic bodies containing the drug have been used to kill tumor cells, with reduced side effects[24]. Cells have been loaded with NPs also— superparamagnetic iron oxide nanoparticles (SPIONs) have been loaded in mesenchymal stem cells to produce charged EVs to treat leukemia[23], while iron oxide NPs and a photosensitizer have been encapsulated in HUVECs and human macrophages to obtain EVs to treat prostate and cervical cancer, respectively [25][26]. Gene therapy can also be carried out with this approach—for example, mesenchymal stem cells have been loaded with different miRNAs to obtain EVs[27]. The purpose of these EVs were varied, i.e., to increase sensitivity of tumor cells to chemotherapeutic drugs (miRNA-122[28]), to inhibit the migration of osteosarcoma cells with miRNA-143[29], and finally to inhibit glioma growth with miRNA-146b[30]. Moreover, chemically modified exogenous mRNA can be loaded in this way into EVs to produce a protein of interest[31].
It is important to focus not only on the technical challenges of producing engineered EVs with indirect methods, but also on the various biological issues that are concerned before, during, and after EV engineering. As a preliminary step before the engineering process, it is important to design the engineered EVs and to make the right choice in terms of parental cells. Many authors decided to use cell lines such as endothelial cell lines (HUVECs) [18][19] or dendritic cells (DCs) [9][12][14], while others worked with more tissue-specific cell lines. From the literature, it is evident that the main challenge in the choice of the parental cells is to become able to work with a patient’s derived cells in a controllable way and with introducing scalable protocols. For example, one of the critical issues is to obtain EVs with characteristics compatible to the cells with which they will interact. During the engineering, it is important to choose the proper surface modification to achieve the purpose and also to pay attention to the possible unwanted effects. Another challenge is to identify the most efficient way to obtain the functionalization. One of the most popular choices is to transfect the parental cells with the right plasmid vectors and their building is nowadays an important investigation subject in the biological field[14][8][9][10][11][12][13][14][21]. The other popular approach is to incubate the cells with DSPE-PEG (1,2-distearoyl-sn-glycero-3-phosphoethanolamine-polyethyleneglycol) to both link and further space the membrane from the targeting molecules. Such functional lipids can be actually bound to targeting ligands such as biotin, folate, thiol groups, or arginylglycylaspartic acid (RGD). Biotin can in turn selectively bind to streptavidin, being used for further functionalization [17][18][19]. Folate is able to target specific cancer cells [16][17], while thiol groups are useful in many binding reactions [32]. RGD is one of the most common sequences of cellular attachment at the extracellular matrix[32].
After the functionalization, the main biological challenge is to choose the most appropriate cell line or animal model to test the engineered EVs. One of the most popular choices is to use immortalized cell lines, for example HeLa [10][32][16], 3T3[17] and Neuro2A [33][8], due to their advantages in terms of cost, ease of use, and ethical concerns. Indeed, even if not specific like the primary cell lines, they allow for the ability to overcome the main biological challenges of EV testing, such as it being time-consuming and having scalability issues, thus allowing movement from in vitro to in vivo testing easily.
Most of the authors that tested their formulation in vivo chose transgenic [11][12] or non-transgenic mice that bear [15][18][19] or do not bear [8][9][13][14][16][17] autologous tumor or xenografts and that could be athymic [20] or not. Unfortunately, these animal models are not complex enough to simulate the human system, and thus more investigation efforts must be pursued to develop more appropriate testing platforms.
3. Direct Methods
Several methods are used to modify the surface of EVs after their isolation. These modifications can be carried out to achieve more specific targeting or mimetic features [6]. Most frequently, the aim is to obtain fluorescent and magnetic labelling to track EVs, their biodistribution, and their pharmacokinetics to investigate their possible diagnostic and therapeutic applications[4]. As EVs are very delicate, it is necessary to pay attention to the reaction conditions to avoid their disruption and aggregation due to inappropriate temperature, pressure, and osmotic stresses[5]. Working in mild conditions can help to obtain the most controlled results[4]. After their isolation, EVs’ surfaces can be modified in different ways, as reported in Table 2 and Figure 3.
Table 2. Applications of the direct methods and graphical abstracts from the references.
|
Parental Cells |
Functionalization |
Functionalization Step |
Recipient Cells |
Treatment Conditions |
Application |
Reference |
|
|
Covalent |
|
||||||
|
PC12 cells |
TAMRA-NHS
|
200 µL of Exos added to 1 mL 0.1 M sodium bicarbonate with 100mg TAMRA-NHS |
PC12 cells |
1×108 cells and 100 µL of exosome solutions |
Visualization of cellular uptake and intracellular trafficking of exosomes |
[34] |
|
|
4T1 cells |
Alkyne groups conjugated with azide-fluor 545 |
80 μg of exosomes in PBS, Cu (II) sulfate pentahydrate, 1.44 M l-ascorbic acid, and bathophenanthrolinedisulfonic acid disodium salt trihydrate |
4T1 cells |
Cells at a confluency of 75% and 5μg of exosomes in 100 μL RPMI |
Surface functionalization of exosomes |
[35] |
|
|
Neuro2A and platelets |
EGFR conjugated to DMPE-PEG derivatives |
Conjugation in a 8.6:1000 molar ratio of nanobody/DMPE-PEG-maleimide micelles and then mixed with EVs |
A431 and Neuro2A IN VIVO: Crl:NU-Foxn1nu mice with human tumor xenografts |
3×104 cells per well and 8 µg/mL of EVs IN VIVO: i.v. of 2.5 µg of EVs in 100 µL PBS |
Enhancing cell specificity and circulation time of EVs |
[36] |
|
|
Bovine serum |
DSPE and chemical conjugation by NHS-PEG |
Physical: DSPE-PEG-biotin mixed with the EXOs (500 µg in PBS) Chemical: NHS-PEG-biotin reacted with the primary amines (500 nmol) on the EXOs |
RAW264.7, DC2.4, and NIH3T3 IN VIVO: mice |
6×105 or 4×105 cells per well and EXOs at an ICG concentration of 5.8 µg per well IN VIVO: s.i. at a DiI dose of 1.52 µg/kg |
Efficient delivery of immune stimulators and antigens to the lymph nodes in vivo |
[37] |
|
|
RAW 264.7 cells and BMM from C57BL/6 mice |
DSPE-PEG or DSPE-PEG-AA |
Addition of DSPE-PEG or DSPE-PEG-AA at 50 μg/mL |
IN VIVO: C57BL/6 with induced pulmonary metastases |
IN VIVO: i.v. injected with the exos at 108 particles/100 μl, n = 4 per group |
Targeted paclitaxel delivery to pulmonary metastases |
[38] |
|
|
HEK293T cells |
FA, PSMA RNA aptamer, and EGFR RNA aptamer conjugated to 3WJ |
Cholesterol-triethylene glycol was conjugated into the arrow-tail of the pRNA-3WJ to promote the anchorage of the 3WJ onto the EV membrane |
MDA-MB-231, KB, LNCaP (PSMA+), PC3 (PSMA–) IN VIVO: KB xenograft mice model |
Incubation with cells IN VIVO: 1 dose of equivalent 0.5 mg siRNA/kg every 3 days for a total of 6 doses |
Control RNA loading and ligand display on EVs for cancer regression |
[39] |
|
|
RAW 264.7 |
NRP-1-targeted peptide RGE |
Surface modification with sulfo-NHS that can react with azide-modified RGE peptide, using salts and copper as catalyst |
U251 and Bel-7404 IN VIVO: orthotopic glioma-bearing BALB/c nude mice |
Cells and exos at the equivalent of 15 µg/mL of Cur/SPIONS IN VIVO: i.v. of Cur/SPIONS at 800 µg/200 µg Exos/200 µL PBS |
Facilitate simultaneous imaging and therapy of glioma in vitro and in vivo |
[40] |
|
|
Non-Covalent |
|
||||||
|
HeLa |
Cationic lipid formulation, LTX, and GALA |
20 μl LTX added to a solution of exosomes and 20 μl GALA and incubated for 20 min at room temperature |
HeLa and (CHO)-K1 |
2 mL with 2 × 105 cells and 20 μg/mL of exosomes |
Enhancing cytosolic delivery of exosomes |
[41] |
|
|
RTCs |
Superparamagnetic magnetite colloidal nanocrystal clusters |
1 mL of serum incubated with 200 µL of M-Tfs solution for 4 h at 4°C |
H22 cells IN VIVO: Kunming mice bearing a subcutaneous H22 cancer |
0.1 mg/mL of exos in a simulated blood circulation at 32.85 cm/s (artery), 14.60 cm/s (vein), and 0.05 cm/s (capillary) |
Targeted drug delivery vehicle for cancer therapy with magnetic properties |
[42] |
|
|
Human serum and C2C12 |
Rhodamine-labelled M12-CP05, FITC-labelled NP41-CP05 |
CP05 (200 µg/mL) incubated with nickel beads, added into the precentrifuged serum (200 µL), and incubated for 30 min at 4°C under rotation |
IN VIVO: dystrophin-deficient and immunodeficient nude mice and C57BL/6 mice |
IN VIVO: i.m.1 or 2 µg of EXOs, i.v. EXOs in 100 µL of saline solution |
Enabling targeting, cargo loading, and capture of exosomes from diverse origins |
[43] |
|
|
4T1, MCF-7, and PC3 |
DiR labelling |
5µL of DIR, at a concentration of 220 µg/mL in ethanol, was mixed with 220 µg exosomes or liposomes in 100 µL PBS for 1 hour |
IN VIVO: Balb/c, nude, and NOD.CB17- Prkdcscid/J mice with either 4T1 cells or PC3 cells |
IN VIVO: i.v. 60 µg DIR-labeled exosomes in 200 µL PBS or i.t. 60 µg of DIR-labeled exosomes in 50 µL PBS |
Biodistribution and delivery efficiency of unmodified tumor-derived exosomes |
[44] |
|
|
Glycosylation |
|
||||||
|
MLP29 |
Neuraminidase |
Surface glycosylation of the EVs was manipulated by treatment with neuraminidase to remove the terminal residues of sialic acid |
IN VIVO: wild-type mice |
IN VIVO: i.v. of the EVs |
Modification of the glycosylation of EVs to alter their biodistribution in vivo |
[45] |
|
|
U87 and GBM8 |
Glycosylation and insertion of targeting ligand to DC-SIGN |
Treated with a pan-sialic acid hydrolase Neuraminidase for 30 min at 37°C and/or incubated with palmitoyl-LewisY while vortexing for 10 min |
MoDCs |
500,000 cells incubated with EVs for 45 min on ice to allow receptor binding |
Enhancing receptor-mediated targeting of dendritic cells |
[46] |
|
|
HEK293FT |
Glycosylation of targeting-peptide-Lamp2b fusion proteins |
1.5 mL of 0.971 M sucrose was slowly pipetted underneath the 8.5 ml of exosome solution |
HEK293FT and Neuro2A |
Cells at 50% confluency and EVs for 2 h at 37 °C |
Stabilization of exosome-targeting peptides |
[47] |
|
|
Hybridization |
|
||||||
|
HEK293FT |
CRISPR/CRISPR‐associated protein 9 (Cas9) system |
Addition of the plasmid–liposome complex to exosomes and incubated at 37 °C for 12 h in a volume ratio of 1:2 |
MSCs |
Incubation with cells at 90% of confluency |
Efficiently encapsulate large plasmids and be endocytosed in MSCs |
[48] |
|
|
RAW 264.7, CMS7-wt, and CMS7-HE |
DOPC, DOPS, DOTAP, and DOPS/PEG-DSPE |
Exosomes (300 μg/mL, protein) mixed with 100 μM liposomes in a volume ratio of 1:1 and then several freeze–thaw cycles |
HeLa cells |
4.5 μg protein in exosome incubated with 1×105 HeLa cells for 4 h at 37 °C |
Control and modify the performance of exosomal nanocarriers |
[49] |
|
|
HUVECs and MSCs |
Phosphatidylcholine, phosphatidylethanolaminein, and PEG |
Liposomes and EVs were mixed at 40 °C in a total volume of 40−200 μL (2×1010 or 2×1011objects); liposome/EV ratio of 1:1, 1:9, or 9:1 in PBS. PEG was added at 5−30% (w/v) |
THP1-derived macrophages and CT26 |
100,000 cells per well and hybrid EVs containing 1 mol % of DiR, cells, and 400 μL of mTHPC-loaded hybrid EVs or (3D) 500 cells and mTHPC-loaded hybrid EVs |
Design of personalized biogenic drug delivery systems |
[50] |
|
|
J774A.1 |
L-a-phosphatidylcholine and cholesterol |
EVs (200 µg protein) used to hydrate the dry 1000 µg of lipid film in a final volume of 1 mL; then, the solution was extruded through 400 and 200 nm polycarbonate membrane filter |
K7M2, 4T1, and NIH/3T3 |
10,000–20,000 cells and 4 mL of 50 µg/mL of hybrid EVs at 37°C for 3h or 48 h |
Tumor targeted drug delivery |
[51] |
|
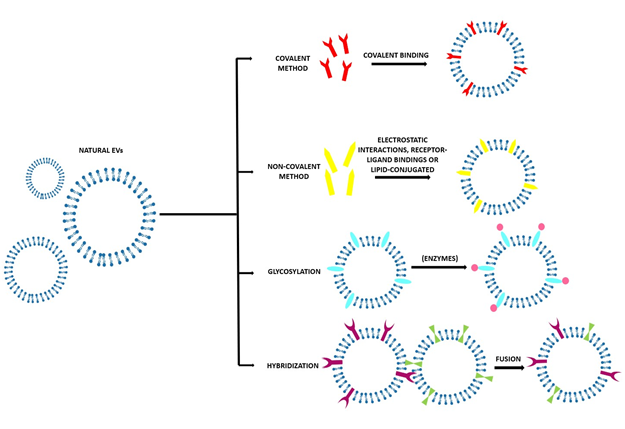
Figure 3. Scheme of the various direct methods to obtain engineered EVs with the desired characteristics and with the molecules of interest on the surface. In particular, covalent method; non-covalent methods such as electrostatic interaction; receptor–ligand binding; or lipid conjugation, glycosylation, or hybridization.
3.1. Covalent Methods
As the classical crosslinking is not enough in terms of specificity and efficiency, the most used covalent method nowadays is the Click Chemistry approach, also known as azide alkyne cycloaddition[52]. With this process, an alkyne moiety reacts with an azide group to form a stable triazole linkage[52]. Some studies also used a copper catalyst to accelerate the reaction[53], but several authors demonstrated that a successful binding can be obtained also without the copper catalyst [54]. One of the strengths of this method is that the experimental conditions are mild and that it can take place in both in organic and aqueous media (water, alcohols, dimethyl sulfoxide (DMSO))[55]. The yield is high, the method is simple, and it does not impact on EV size nor on the target cell uptake[55]. This method does have, however, some drawbacks—the alkyne modification of the EV surface most likely occurs on the amine groups of the proteins instead of those of the phospholipids, introducing the possibility that the EV protein function may be inhibited [52]. By controlling the number of alkyne groups, it is possible to avoid the over modification of EV membrane proteins—with a standard calibration curve it has been estimated that approximately 1.5 alkyne modifications are made for every 150 kDa of EV protein[35]. A very common approach is PEGylation, the modification of EVs’ surfaces with polyethylene glycol to extend the circulation half time of the EVs. The drawback of PEGylation is that the PEG corona also reduces the EV–cell interaction and the cellular uptake of the EVs[36]. This disadvantage can be overcome by functionalizing the distal end of the PEG chain with a targeting ligand[4].
3.2. Non-Covalent Methods
These methods are based on mild reactions, such as electrostatic interactions, receptor–ligand bindings, and lipid-conjugated compounds post-insertion into the EV’s lipid bilayer[4]. Electrostatic approaches usually involve highly cationic species adhering on negatively charged functional groups present on the biological membranes [4]. A possible drawback of these methods is that certain cationic nanomaterials can cause cytotoxicity and that they are typically taken up into the cells via endocytosis, leading to lysosomal degradation[5].
3.3. Glycosylation
Glycosylation is at the base of many biological functions of EVs, such as cargo protein recruitment and cellular recognition and uptake [56][57]. Alterations in the glycosylation pattern has been associated with different pathologies, for example, cancer, and these changes are closely correlated with the specific malignant transformation and progression. This evidence has led to make glycan structure a useful target for anti-tumor applications in theranostics [58][59]. The manipulation of glycosylation can be done using either enzymes or not.
3.4. Hybridization
This method implies the fusion of natural EVs with their artificial counterpart, liposomes, to optimize the properties of native EVs [26]. This can be obtained thanks to the lipid composition of the EV membrane. In this way, the colloidal stability of EVs is improved, increasing their half-life in blood and modifying their immunogenicity profile, possibly decreasing it[52]. The lipid composition has been evidenced to impact on the cellular uptake—EVs hybridized with neutral or anionic lipids have a higher possibility to be taken up by cells than those hybridized with cationic lipids[52]. Moreover, hybridization of EVs increases the vesicle size (in a technique-dependent way)—this is a drawback because it decreases the in vivo retention of the vesicles, but also an advantage as it can improve the drug encapsulation efficiency[52]. Native EVs are actually very small in size and thus limited in their ability to encapsulate large molecules, while larger hybridized EVs can carry larger cargos[52].
As for the indirect methods, it is important to remember that the technical challenges to engineer the EVs with the different direct methods are directly correlated with the biological challenges that are fundamental in every step of EV engineering, from the preliminary design to the real environment testing. For what concerns the choice of the parental cells, in some works the authors chose the RAW 264.7 macrophages, an immortalized cancer cell line [38][40][49], while others used immortalized cell lines such as HeLa [41] or Neuro 2A[36], or even extracted the desired cells directly from mice[38] or human serum[43]. As stated previously, the main biological challenge is to find a scalable and controllable way to use the patient’s cells as source in order to obtain EVs that are possibly compatible to the patient environment.
Moreover, the best EV engineering method must be carefully evaluated in a specific context, considering advantages and limitations. In particular, for what concerns the functionalization, it is important to find the proper molecule for the desired purpose, and a variety of functionalizations are reported in the literature, as mentioned above. As for the indirect methods, the use of DSPE-PEG [37][38][49] or DMPE-PEG[36], as spacer to expose the functionalization, is a commonly used strategy. Finally, for both in vitro and in vivo testing steps, the biological challenges are the same listed above and analyzed for the indirect methods in terms of choice of the best cell line and/or animal model.
At this point, it is clear that the functionalization of EVs with ligands and other molecules can boost up their stability in blood circulation, have the capability of localizing the target site, and can increase their intracellular delivery efficiency[60]. The main drawback of EV engineering is the introduction of the risks of altering the orientation of membrane proteins, which may compromise their biological functionalities or even induce immunogenicity[6]. Further risks of EV engineering are associated with the hiding of these proteins or to the damage or disruption the EV membrane[50].
References
- Clotilde Théry; Kenneth W. Witwer; Elena Aikawa; Maria Jose Alcaraz; Johnathon D Anderson; Ramaroson Andriantsitohaina; Anna Antoniou; Tanina Arab; Fabienne Archer; Georgia K Atkin-Smith; et al.D Craig AyreJean-Marie BachDaniel BachurskiHossein BaharvandLeonora BalajShawn BaldacchinoNatalie N BauerAmy A BaxterMary BebawyCarla BeckhamApolonija Bedina ZavecAbderrahim BenmoussaA C BerardiPaolo BergeseEwa BielskaCherie BlenkironSylwia Bobis-WozowiczEric BoilardWilfrid BoireauAntonella BongiovanniFrancesc E. BorràsSteffi BöschC. M. BoulangerXandra BreakefieldAndrew M BreglioMeadhbh Á BrennanDavid R BrigstockAlain BrissonMarike Ld BroekmanJacqueline F BrombergPaulina Bryl-GóreckaShilpa BuchAmy H BuckDylan BurgerSara BusattoDominik BuschmannBenedetta BussolatiEdit I BuzásJ. Brian ByrdGiovanni CamussiDavid Rf CarterSarah CarusoLawrence W. ChamleyYu-Ting ChangChihchen ChenShuai ChenLesley ChengAndrew R ChinAled ClaytonStefano P ClericiAlex CocksEmanuele CocucciRobert J CoffeyAnabela Cordeiro-Da-SilvaYvonne CouchFrank Aw CoumansBeth CoyleRossella CrescitelliMiria Ferreira CriadoCrislyn D’Souza-SchoreySaumya DasAmrita Datta ChaudhuriPaola De CandiaEliezer F De SantanaOlivier De WeverHernando A Del PortilloTanguy DemaretSarah DevilleA. DevittBert DhondtDolores Di VizioLothar C DieterichVincenza DoloAna Paula Dominguez RubioMassimo DominiciMauricio R DouradoTom Ap DriedonksFilipe DuarteHeather M DuncanRamon M. EichenbergerKarin EkströmSamir El AndaloussiCeline Elie-CailleUta ErdbrüggerJuan M Falcón-PérezFarah FatimaJason E. FishMiguel Flores-BellverAndrás FörsönitsAnnie Frelet-BarrandFabia FrickeGregor FuhrmannSusanne GabrielssonAna Gámez-ValeroChris GardinerKathrin GärtnerRaphael GaudinYong Song GhoBernd GiebelCaroline GilbertMario GimonaIlaria GiustiDeborah Ci GoberdhanAndre GoergensSharon M. GorskiDavid W. GreeningJulia Christina GrossAlice GualerziGopal N GuptaDakota GustafsonAase HandbergReka A HarasztiPaul HarrisonHargita HegyesiAn HendrixAndrew F. HillFred H HochbergKarl F HoffmannBeth HolderHarry HolthoferBaharak HosseinkhaniGuoku HuYiyao HuangVeronica HuberStuart HuntAhmed Gamal-Eldin IbrahimTsuneya IkezuJameel InalMustafa IşınAlena IvanovaHannah K JacksonSøren JacobsenSteven M JayMuthuvel JayachandranGuido JensterLanzhou JiangSuzanne M. JohnsonJennifer C. JonesAmbrose JongTijana Jovanovic-TalismanStephanie JungRaghu KalluriShin-Ichi KanoSukhbir KaurYumi KawamuraEvan T KellerDelaram KhamariElena KhomyakovaAnastasia KhvorovaPeter KierulfKwang Pyo KimThomas KislingerMikael KlingebornDavid J KlinkeMiroslaw KornekMaja M. KosanovićÁrpád Ferenc KovácsEva-Maria Krämer-AlbersSusanne KrasemannMirja KrauseIgor V KurochkinGina D KusumaSören KuypersSaara LaitinenScott M. LangevinLucia R. LanguinoJoanne LanniganCecilia LässerLouise C LaurentGregory LavieuElisa Lazaro-IbanezSoazig Le LayMyung-Shin LeeYi Xin Fiona LeeDebora S LemosMetka LenassiAleksandra LeszczynskaIsaac Ts LiKe LiaoSten F LibregtsErzsebet LigetiRebecca LimSai Kiang LimAija LinēKaren LinnemannstönsAlicia LlorenteCatherine A LombardMagdalena J LorenowiczÁkos M. LőrinczJan LötvallJason LovettMichelle C LowryXavier LoyerQuan LuBarbara LukomskaTaral R LunavatSybren Ln MaasHarmeet MalhiAntonio MarcillaJacopo MarianiJavier MariscalElena Martens-UzunovaLorena Martín-JaularM Carmen MartinezVilma R MartinsMathilde MathieuSuresh MathivananMarco MaugeriLynda K McGinnisMark J McVeyDavid G MeckesKatie L MeehanInge MertensValentina R MinciacchiAndreas MöllerMalene Møller JørgensenAizea Morales-KastresanaJess MorhayimFrançois MullierMaurizio MuracaL. MusanteVeronika MussackDillon C MuthK H MyburghTanbir NajranaMuhammad NawazIrina NazarenkoPeter NejsumChristian NeriTommaso NeriRienk Nieuwland2Leonardo NimrichterJohn P NolanEsther Nm Nolte-’T HoenNicole Noren HootenLorraine O’DriscollTina O’GradyAna O'loghlenTakahiro OchiyaMartin OlivierAlberto OrtizLuis A. OrtizXabier OsteikoetxeaOle ØstergaardMatias OstrowskiJaesung ParkD. Michiel PegtelHéctor PeinadoFrancesca PerutMichael W. PfafflDonald G. PhinneyBartijn Ch PietersRyan PinkDavid S PisetskyElke Pogge Von StrandmannIva PolakovicovaIvan K. PoonBonita H PowellIlaria PradaLynn PulliamPeter QuesenberryAnnalisa RadeghieriRobert L RaffaiStefania RaimondoJanusz RakMarcel I RamirezGraça RaposoMorsi S RayyanNeta Regev-RudzkiFranz L RicklefsPaul D RobbinsDavid D. RobertsSilvia C RodriguesEva RohdeSophie RomeKasper Ma RouschopAurelia RughettiAshley E RussellPaula SaáSusmita SahooEdison Salas-HuenuleoCatherine SánchezJulie A. SaugstadMeike J SaulRaymond SchiffelersRaphael SchneiderTine Hiorth SchøyenAaron ScottEriomina ShahajShivani SharmaOlga ShatnyevaFaezeh ShekariGanesh Vilas ShelkeAshok K. ShettyKiyotaka Shiba Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. Journal of Extracellular Vesicles 2018, 7, 1535750, 10.1080/20013078.2018.1535750.
- Akhil Srivastava; Narsireddy Amreddy; Vipul Pareek; Mahendran Chinnappan; Rebaz Ahmed; Meghna Mehta; Mohammad Razaq; Anupama Munshi; Rajagopal Ramesh; Progress in extracellular vesicle biology and their application in cancer medicine. WIREs Nanomedicine and Nanobiotechnology 2020, 12, 4, 10.1002/wnan.1621.
- Sarah Caruso; Ivan K. Poon; Apoptotic Cell-Derived Extracellular Vesicles: More Than Just Debris. Frontiers in Immunology 2018, 9, 1486, 10.3389/fimmu.2018.01486.
- Francesca Susa; Tania Limongi; Bianca Dumontel; Veronica Vighetto; Valentina Cauda; Engineered Extracellular Vesicles as a Reliable Tool in Cancer Nanomedicine. Cancers 2019, 11, 1979, 10.3390/cancers11121979.
- Sophia G. Antimisiaris; Spyridon Mourtas; Antonia Marazioti; Exosomes and Exosome-Inspired Vesicles for Targeted Drug Delivery. Pharmaceutics 2018, 10, 218, 10.3390/pharmaceutics10040218.
- Kyle I. Mentkowski; Jonathan D. Snitzer; Sarah Rusnak; Jennifer K. Lang; Therapeutic Potential of Engineered Extracellular Vesicles. The AAPS Journal 2018, 20, 50, 10.1208/s12248-018-0211-z.
- Zachary Stickney; Joseph Losacco; Sophie McDevitt; Zhiwen J Zhang; Biao Lu; Development of exosome surface display technology in living human cells. Biochemical and Biophysical Research Communications 2016, 472, 53-59, 10.1016/j.bbrc.2016.02.058.
- Lydia Alvarez-Erviti; Yiqi Seow; Haifang Yin; Corinne Betts; Samira Lakhal; Matthew J. A. Wood; Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nature Biotechnology 2011, 29, 341-345, 10.1038/nbt.1807.
- Yanhua Tian; Suping Li; Jian Song; Tianjiao Ji; Motao Zhu; Gregory J. Anderson; Jingyan Wei; Guangjun Nie; A doxorubicin delivery platform using engineered natural membrane vesicle exosomes for targeted tumor therapy. Biomaterials 2014, 35, 2383-2390, 10.1016/j.biomaterials.2013.11.083.
- Sander A. A. Kooijmans; Clara Gómez Aleza; Steve R. Roffler; Wouter Van Solinge; Pieter Vader; Raymond Schiffelers; Display of GPI-anchored anti-EGFR nanobodies on extracellular vesicles promotes tumour cell targeting. Journal of Extracellular Vesicles 2016, 5, 31053, 10.3402/jev.v5.31053.
- Shin-Ichiro Ohno; Masakatsu Takanashi; Katsuko Sudo; Shinobu Ueda; Akio Ishikawa; Nagahisa Matsuyama; Koji Fujita; Takayuki Mizutani; Tadaaki Ohgi; Takahiro Ochiya; et al.Noriko GotohMasahiko Kuroda Systemically Injected Exosomes Targeted to EGFR Deliver Antitumor MicroRNA to Breast Cancer Cells. Molecular Therapy 2012, 21, 185-191, 10.1038/mt.2012.180.
- Zachary C. Hartman; Junping Wei; Oliver K. Glass; Hongtao Guo; Gangjun Lei; Xiao-Yi Yang; Takuya Osada; Amy Hobeika; Alain Delcayre; Jean-Bernard Le Pecq; et al.Michael A. MorseTimothy M. ClayHerbert Kim Lyerly Increasing vaccine potency through exosome antigen targeting. Vaccine 2011, 29, 9361-9367, 10.1016/j.vaccine.2011.09.133.
- R. B. Rountree; S. J. Mandl; J. M. Nachtwey; K. Dalpozzo; L. Do; J. R. Lombardo; P. L. Schoonmaker; K. Brinkmann; U. Dirmeier; R. Laus; et al.A. Delcayre Exosome Targeting of Tumor Antigens Expressed by Cancer Vaccines Can Improve Antigen Immunogenicity and Therapeutic Efficacy. Cancer Research 2011, 71, 5235-5244, 10.1158/0008-5472.can-10-4076.
- Alain Delcayre; Angeles Estellés; Jeffrey Sperinde; Thibaut Roulon; Pedro Paz; Barbara Aguilar; Janeth Villanueva; Susu Khine; Jean-Bernard Le Pecq; Exosome Display technology: Applications to the development of new diagnostics and therapeutics. Blood Cells, Molecules, and Diseases 2005, 35, 158-168, 10.1016/j.bcmd.2005.07.003.
- Jie Wang; Yue Dong; Yiwei Li; Wei Li; Kai Cheng; Yuan Qian; Guoqiang Xu; Xiaoshuai Zhang; Liang Hu; Peng Chen; et al.Wei DuXiaojun FengYuan-Di ZhaoZhihong ZhangBi-Feng Liu Designer Exosomes for Active Targeted Chemo-Photothermal Synergistic Tumor Therapy. Advanced Functional Materials 2018, 28, 1707360, 10.1002/adfm.201707360.
- Wei Zhang; Zi-Li Yu; Min Wu; Jian-Gang Ren; Hou-Fu Xia; Guo-Liang Sa; Jun-Yi Zhu; Dai-Wen Pang; Yi-Fang Zhao; Gang Chen; et al. Magnetic and Folate Functionalization Enables Rapid Isolation and Enhanced Tumor-Targeting of Cell-Derived Microvesicles. ACS Nano 2017, 11, 277-290, 10.1021/acsnano.6b05630.
- Lian Zhu; Di Dong; Zi-Li Yu; Yi-Fang Zhao; Dai-Wen Pang; Zhi-Ling Zhang; Folate-Engineered Microvesicles for Enhanced Target and Synergistic Therapy toward Breast Cancer. ACS Applied Materials & Interfaces 2017, 9, 5100-5108, 10.1021/acsami.6b14633.
- Gang Chen; Jun-Yi Zhu; Zhi-Ling Zhang; Wei Zhang; Jian-Gang Ren; Min Wu; Zheng-Yuan Hong; Cheng Lv; Dai-Wen Pang; Yi-Fang Zhao; et al. Transformation of Cell-Derived Microparticles into Quantum-Dot-Labeled Nanovectors for Antitumor siRNA Delivery. Angewandte Chemie International Edition 2014, 54, 1036-1040, 10.1002/anie.201410223.
- Jie Wang; Wei Li; Leicheng Zhang; Lin Ban; Peng Chen; Wei Du; Xiaojun Feng; Bi-Feng Liu; Chemically Edited Exosomes with Dual Ligand Purified by Microfluidic Device for Active Targeted Drug Delivery to Tumor Cells. ACS Applied Materials & Interfaces 2017, 9, 27441-27452, 10.1021/acsami.7b06464.
- Charles Pin-Kuang Lai; Osama Mardini; Maria Ericsson; Shilpa Prabhakar; Casey A. Maguire; John W. Chen; Bakhos A. Tannous; Xandra O. Breakefield; Dynamic Biodistribution of Extracellular Vesicles in Vivo Using a Multimodal Imaging Reporter. ACS Nano 2014, 8, 483-494, 10.1021/nn404945r.
- Masaki Morishita; Yuki Takahashi; Makiya Nishikawa; Reiichi Ariizumi; Yoshinobu Takakura; Enhanced Class I Tumor Antigen Presentation via Cytosolic Delivery of Exosomal Cargos by Tumor-Cell-Derived Exosomes Displaying a pH-Sensitive Fusogenic Peptide. Molecular Pharmaceutics 2017, 14, 4079-4086, 10.1021/acs.molpharmaceut.7b00760.
- Luisa Pascucci; Valentina Coccè; Arianna Bonomi; Diletta Ami; Piero Ceccarelli; Emilio Ciusani; Lucia Viganò; Alberta Locatelli; Francesca Sisto; Silvia Maria Doglia; et al.Eugenio ParatiMaria Ester BernardoMaurizio MuracaGiulio AlessandriGianpietro BondiolottiAugusto Pessina Paclitaxel is incorporated by mesenchymal stromal cells and released in exosomes that inhibit in vitro tumor growth: A new approach for drug delivery. Journal of Controlled Release 2014, 192, 262-270, 10.1016/j.jconrel.2014.07.042.
- Chao Liu; Haiyan Gao; Peng Lv; Jingyi Liu; Gang Liu; Extracellular vesicles as an efficient nanoplatform for the delivery of therapeutics. Human Vaccines & Immunotherapeutics 2017, 13, 2678-2687, 10.1080/21645515.2017.1363935.
- Ke Tang; Yi Zhang; Huafeng Zhang; Pingwei Xu; Jing Liu; Jingwei Ma; Meng Lv; Dapeng Li; Foad Katirai; Guan-Xin Shen; et al.Guimei ZhangZuo-Hua FengDuyun YeBo Huang Delivery of chemotherapeutic drugs in tumour cell-derived microparticles. Nature Communications 2012, 3, 1282, 10.1038/ncomms2282.
- Bárbara Mulén; Alba Nicolas-Boluda; Amanda K Andriola Silva; Florence Gazeau; Theranostic Iron Oxide Nanoparticle Cargo Defines Extracellular Vesicle-Dependent Modulation of Macrophage Activation and Migratory Behavior. Advanced Biosystems 2018, 2, 1800079, 10.1002/adbi.201800079.
- Amanda K.A. Silva; Jelena Kolosnjaj-Tabi; Stephanie Bonneau; Iris Marangon; Nicole Boggetto; Kelly Aubertin; Olivier Clément; Michel Francis Bureau; Nathalie Luciani; Florence Gazeau; et al.Claire Wilhelm Magnetic and Photoresponsive Theranosomes: Translating Cell-Released Vesicles into Smart Nanovectors for Cancer Therapy. ACS Nano 2013, 7, 4954-4966, 10.1021/nn400269x.
- Max Piffoux; Amanda K.A. Silva; Jean-Baptiste Lugagne; Pascal Hersen; Claire Wilhelm; Florence Gazeau; Extracellular Vesicle Production Loaded with Nanoparticles and Drugs in a Trade-off between Loading, Yield and Purity: Towards a Personalized Drug Delivery System. Advanced Biosystems 2017, 1, 1700044, 10.1002/adbi.201700044.
- Guohua Lou; Xiuli Song; Fan Yang; Shanshan Wu; Jing Wang; Zhi Chen; Yanning Liu; Exosomes derived from miR-122-modified adipose tissue-derived MSCs increase chemosensitivity of hepatocellular carcinoma.. Journal of Hematology & Oncology 2015, 8, 122, 10.1186/s13045-015-0220-7.
- Keisuke Shimbo; Shigeru Miyaki; Hiroyuki Ishitobi; Yoshio Kato; Tadahiko Kubo; Shoji Shimose; Mitsuo Ochi; Exosome-formed synthetic microRNA-143 is transferred to osteosarcoma cells and inhibits their migration. Biochemical and Biophysical Research Communications 2014, 445, 381-387, 10.1016/j.bbrc.2014.02.007.
- Mark Katakowski; Ben Buller; Xuguang Zheng; Yong Lu; Thomas Rogers; Oyinkansola Osobamiro; Wayne Shu; Feng Jiang; Michael Chopp; Exosomes from marrow stromal cells expressing miR-146b inhibit glioma growth.. Cancer Letters 2013, 335, 201-4, 10.1016/j.canlet.2013.02.019.
- Marco Maugeri; Muhammad Nawaz; Alexandros Papadimitriou; Annelie Angerfors; Alessandro Camponeschi; Manli Na; Mikko Hölttä; Pia Skantze; Svante Johansson; Martina Sundqvist; et al.Johnny LindquistTomas KjellmanInga-Lill MårtenssonTao JinPer SunnerhagenSofia ÖstmanLennart LindforsHadi Valadi Linkage between endosomal escape of LNP-mRNA and loading into EVs for transport to other cells. Nature Communications 2019, 10, 4333-15, 10.1038/s41467-019-12275-6.
- Jie Wang; Yue Dong; Yiwei Li; Wei Li; Kai Cheng; Yuan Qian; Guoqiang Xu; Xiaoshuai Zhang; Liang Hu; Peng Chen; et al.Wei DuXiaojun FengYuan-Di ZhaoZhihong ZhangBi-Feng Liu Designer Exosomes for Active Targeted Chemo-Photothermal Synergistic Tumor Therapy. Advanced Functional Materials 2018, 28, 277-90, 10.1002/adfm.201707360.
- MikoŁaj P. Zaborowski; Leonora Balaj; Xandra O. Breakefield; Charles Pin-Kuang Lai; Extracellular Vesicles: Composition, Biological Relevance, and Methods of Study. BioScience 2015, 65, 783-797, 10.1093/biosci/biv084.
- Tian Tian; Yuanyuan Wang; Haitao Wang; Zhaoqi Zhu; Zhongdang Xiao; Visualizing of the cellular uptake and intracellular trafficking of exosomes by live-cell microscopy. Journal of Cellular Biochemistry 2010, 111, 488-496, 10.1002/jcb.22733.
- Tyson Smyth; Krastina Petrova; Nicole M. Payton; Indushekhar Persaud; Jasmina S. Redzic; Michael W. Graner; Peter Smith-Jones; Thomas J. Anchordoquy; Surface Functionalization of Exosomes Using Click Chemistry. Bioconjugate Chemistry 2014, 25, 1777-1784, 10.1021/bc500291r.
- S.A.A. Kooijmans; L.A.L. Fliervoet; R. Van Der Meel; M.H.A.M. Fens; H.F.G. Heijnen; P.M.P. Van Bergen En Henegouwen; Pieter Vader; Raymond Schiffelers; PEGylated and targeted extracellular vesicles display enhanced cell specificity and circulation time. Journal of Controlled Release 2016, 224, 77-85, 10.1016/j.jconrel.2016.01.009.
- Eun Seo Choi; Jihyeon Song; Yoon Young Kang; Hyejung Mok; Mannose-Modified Serum Exosomes for the Elevated Uptake to Murine Dendritic Cells and Lymphatic Accumulation.. Macromolecular Bioscience 2019, 19, e1900042, 10.1002/mabi.201900042.
- Myung Soo Kim; Matthew J. Haney; Yuling Zhao; Dongfen Yuan; Irina Deygen; Natalia L. Klyachko; Alexander V. Kabanov; Elena V. Batrakova; Engineering macrophage-derived exosomes for targeted paclitaxel delivery to pulmonary metastases: in vitro and in vivo evaluations. Nanomedicine: Nanotechnology, Biology and Medicine 2018, 14, 195-204, 10.1016/j.nano.2017.09.011.
- Fengmei Pi; Daniel W. Binzel; Tae Jin Lee; Zhefeng Li; Meiyan Sun; Piotr Rychahou; Hui Li; Farzin Haque; Shaoying Wang; Carlo M. Croce; et al.Bin GuoB. Mark EversPeixuan Guo Nanoparticle orientation to control RNA loading and ligand display on extracellular vesicles for cancer regression. Nature Nanotechnology 2017, 13, 82-89, 10.1038/s41565-017-0012-z.
- Gang Jia; Yong Han; Yanli An; Yinan Ding; Chen He; Xihui Wang; Qiusha Tang; NRP-1 targeted and cargo-loaded exosomes facilitate simultaneous imaging and therapy of glioma in vitro and in vivo. Biomaterials 2018, 178, 302-316, 10.1016/j.biomaterials.2018.06.029.
- Ikuhiko Nakase; Shiroh Futaki; Combined treatment with a pH-sensitive fusogenic peptide and cationic lipids achieves enhanced cytosolic delivery of exosomes. Scientific Reports 2015, 5, 10112, 10.1038/srep10112.
- Hongzhao Qi; Chaoyong Liu; Lixia Long; Yu Ren; Shanshan Zhang; Xiaodan Chang; Xiaomin Qian; Huanhuan Jia; Jin Zhao; Jinjin Sun; et al.Xin HouXubo YuanChun-Sheng Kang Blood Exosomes Endowed with Magnetic and Targeting Properties for Cancer Therapy. ACS Nano 2016, 10, 3323-3333, 10.1021/acsnano.5b06939.
- Xianjun Gao; Ning Ran; Xue Dong; Bingfeng Zuo; Rong Yang; Qibing Zhou; Hong M. Moulton; Yiqi Seow; Haifang Yin; Anchor peptide captures, targets, and loads exosomes of diverse origins for diagnostics and therapy. Science Translational Medicine 2018, 10, eaat0195, 10.1126/scitranslmed.aat0195.
- Tyson Smyth; Max Kullberg; Noeen Malik; Peter Smith-Jones; Michael W. Graner; Thomas J. Anchordoquy; Biodistribution and Delivery Efficiency of Unmodified Tumor-Derived Exosomes. Journal of Controlled Release 2014, 199, 145-155, 10.1016/j.jconrel.2014.12.013.
- Felix Royo; Unai Cossío; Ane Ruiz De Angulo; Jordi Llop; Juan Falcón-Pérez; Modification of the glycosylation of extracellular vesicles alters their biodistribution in mice. Nanoscale 2019, 11, 1531-1537, 10.1039/c8nr03900c.
- Sophie A. Dusoswa; Sophie K. Horrevorts; Martino Ambrosini; Hakan Kalay; Nanne J. Paauw; Rienk Nieuwland; Michiel D. Pegtel; Tom Würdinger; Yvette Van Kooyk; Juan J. Garcia-Vallejo; et al. Glycan modification of glioblastoma-derived extracellular vesicles enhances receptor-mediated targeting of dendritic cells. Journal of Extracellular Vesicles 2019, 8, 1648995, 10.1080/20013078.2019.1648995.
- Michelle Hung; Joshua N. Leonard; Stabilization of Exosome-targeting Peptides via Engineered Glycosylation*. Journal of Biological Chemistry 2015, 290, 8166-8172, 10.1074/jbc.M114.621383.
- Yao Lin; Jiahua Wu; Weihuai Gu; Yulei Huang; Zhongchun Tong; Lijia Huang; Jiali Tan; Exosome-Liposome Hybrid Nanoparticles Deliver CRISPR/Cas9 System in MSCs. Advanced Science 2018, 5, 1700611, 10.1002/advs.201700611.
- Yuko T. Sato; Kaori Umezaki; Shinichi Sawada; Sada-Atsu Mukai; Yoshihiro Sasaki; Naozumi Harada; Hiroshi Shiku; Kazunari Akiyoshi; Engineering hybrid exosomes by membrane fusion with liposomes. Scientific Reports 2016, 6, 21933, 10.1038/srep21933.
- Max Piffoux; Amanda K.A. Silva; Claire Wilhelm; Florence Gazeau; David Tareste; Modification of Extracellular Vesicles by Fusion with Liposomes for the Design of Personalized Biogenic Drug Delivery Systems. ACS Nano 2018, 12, 6830-6842, 10.1021/acsnano.8b02053.
- Sagar Rayamajhi; Tuyen Duong Thanh Nguyen; Ramesh Marasini; Santosh Aryal; Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomaterialia 2019, 94, 482-494, 10.1016/j.actbio.2019.05.054.
- Lalithasri Ramasubramanian; Priyadarsini Kumar; Aijun Wang; Engineering Extracellular Vesicles as Nanotherapeutics for Regenerative Medicine. Biomolecules 2019, 10, 48, 10.3390/biom10010048.
- Stanislav I. Presolski; Vu Phong Hong; M. G. Finn; Copper‐Catalyzed Azide–Alkyne Click Chemistry for Bioconjugation. Current Protocols in Chemical Biology 2011, 3, 153-162, 10.1002/9780470559277.ch110148.
- John C. Jewett; Carolyn R. Bertozzi; Cu-free click cycloaddition reactions in chemical biology.. Chem. Soc. Rev. 2010, 39, 1272-9, 10.1039/b901970g.
- Mauro Cataldi; Chiara Vigliotti; Teresa Mosca; Mariarosaria Cammarota; Domenico Capone; Emerging Role of the Spleen in the Pharmacokinetics of Monoclonal Antibodies, Nanoparticles and Exosomes. International Journal of Molecular Sciences 2017, 18, 1249, 10.3390/ijms18061249.
- Yaxuan Liang; William S. Eng; David R. Colquhoun; Rhoel R. Dinglasan; David R. Graham; Lara K. Mahal; ComplexN-Linked Glycans Serve as a Determinant for Exosome/Microvesicle Cargo Recruitment. Journal of Biological Chemistry 2014, 289, 32526-32537, 10.1074/jbc.m114.606269.
- Charles Williams; Raquel Pazos; Félix Royo; Esperanza González; Meritxell Roura-Ferrer; Aitor Martinez; Jorge Gamiz; Niels-Christian Reichardt; Juan M Falcón-Pérez; Assessing the role of surface glycans of extracellular vesicles on cellular uptake. Scientific Reports 2019, 9, 1-14, 10.1038/s41598-019-48499-1.
- Lulu Cai; Zhipeng Gu; Jian Zhong; Di Wen; Guojun Chen; Lin He; Jun Wu; Zhen Gu; Advances in glycosylation-mediated cancer-targeted drug delivery. Drug Discovery Today 2018, 23, 1126-1138, 10.1016/j.drudis.2018.02.009.
- Stefan Mereiter; Meritxell Balmaña; D. Campos; Joana Gomes; Celso A Reis; Glycosylation in the Era of Cancer-Targeted Therapy: Where Are We Heading?. Cancer Cell 2019, 36, 6-16, 10.1016/j.ccell.2019.06.006.
- Mei Lu; Yuanyu Huang; Bioinspired exosome-like therapeutics and delivery nanoplatforms. Biomaterials 2020, 242, 119925, 10.1016/j.biomaterials.2020.119925.


