Your browser does not fully support modern features. Please upgrade for a smoother experience.
Submitted Successfully!
 +1 credit
+1 credit
Thank you for your contribution! You can also upload a video entry or images related to this topic.
For video creation, please contact our Academic Video Service.
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Wenbin Jia | + 1954 word(s) | 1954 | 2021-05-27 10:39:43 | | | |
| 2 | Catherine Yang | Meta information modification | 1954 | 2021-06-03 10:51:38 | | | | |
| 3 | Catherine Yang | Meta information modification | 1954 | 2021-06-03 10:52:00 | | |
Video Upload Options
We provide professional Academic Video Service to translate complex research into visually appealing presentations. Would you like to try it?
Cite
If you have any further questions, please contact Encyclopedia Editorial Office.
Jia, W. Nicotine-Induced Addiction. Encyclopedia. Available online: https://encyclopedia.pub/entry/10463 (accessed on 08 February 2026).
Jia W. Nicotine-Induced Addiction. Encyclopedia. Available at: https://encyclopedia.pub/entry/10463. Accessed February 08, 2026.
Jia, Wenbin. "Nicotine-Induced Addiction" Encyclopedia, https://encyclopedia.pub/entry/10463 (accessed February 08, 2026).
Jia, W. (2021, June 03). Nicotine-Induced Addiction. In Encyclopedia. https://encyclopedia.pub/entry/10463
Jia, Wenbin. "Nicotine-Induced Addiction." Encyclopedia. Web. 03 June, 2021.
Copy Citation
Nicotine is the predominant addictive compound of tobacco and causes the acquisition of dependence through its interactions with nicotinic acetylcholine receptors and various neurotransmitter releases in the central nervous system.
nicotine-induced addiction
Ca2+/calmodulin-dependent protein kinase II
extracellular signal-regulated kinase
1. Introduction
Smoking was the second leading cause of early death and disability in 2015 [1]. It caused approximately 6.4 million deaths, which accounted for 11.5% of all deaths worldwide, and led to 150 million people becoming disabled in 2015 [2]. In developed countries, inhalation of smoking is considered the largest single cause of early death. Smoking is the reason for 20% of all premature deaths and more than one-third of all deaths in men aged 35–69 years [3]. Accumulated evidence suggests that regular smoking can enhance the risk of coronary heart disease, chronic obstructive pulmonary disease, and cancers of the upper aerodigestive tract and lung [4]. Nicotine in tobacco is considered the predominant addictive component, and it causes continued use in humans [5][6][7]. Nicotine-induced addiction is reported to be difficult to quit. More than 80% of attempts to quit fail within a year, and some highly addicted smokers are able to quit smoking only for a few hours [8][9]. Therefore, people are inclined to use tobacco continually despite the harmful consequences. Other psychostimulant drugs, such as morphine, cocaine, and amphetamine, are similar to nicotine: they participate and modulate the brain reward system and motivation though they have different interaction sites and chemical structures [10].
2. CaMKII in Nicotine and Other Psychostimulant-Induced Addiction
CaMKII plays a critical role in the molecular pathway of the reward system in drug addiction, including nicotine dependence, and affects animal responses to drug abuse [11]. Previous investigations demonstrated that 14 or 28 consecutive days of chronic nicotine administration significantly increased CPP scores in conditioning, nicotine withdrawal, as well as relapse phases and CaMKII autophosphorylation levels in the mouse NAc and hippocampal CA1 region [12]. The number of CaMKII autophosphorylation-positive cells in the NAc was also elevated following chronic nicotine administration relative to saline-administered mice [12]. Intracerebroventricular infusion of CaMKII antagonists KN-62 and KN-93 into mice showed impaired nicotine-induced CPP [13]. CaMKIIα heterozygous (+/−) mice failed to show nicotine-induced CPP behavior, and the increased CaMKII activity in the NAc and VTA in wild-type (WT) mice were blocked by the administration of KN-62 [13]. Nicotine induces significant CPP and elevation of CaMKII activity in a concentration-dependent manner. Some studies identified that intraperitoneal injection of nicotine at doses of 0.25 mg/kg and 1.0 mg/kg could not produce nicotine-induced conditioning, whereas that at a dose of 0.5 mg/kg could [14][15]. The concentration at 2 mg/kg induced conditioned place aversion but not CPP in mice [14]. Nicotine concentration lower than 0.1 mg/kg or higher than 1.4 mg/kg could not induce CPP in subcutaneously injected rats [16]. Although different neurochemical outcomes can be observed with different concentrations of nicotine, our previous data suggest that the daily dose of nicotine at 0.5 mg/kg could execute its functions that induce significant CPP, and elevate CaMKII autophosphorylation level as well as the phosphorylation level of its downstream targets in the NAc and hippocampal CA1 region using an immunoblotting technique [12]. Consistent with nicotine, other addictive drugs such as morphine and methamphetamine have a similar effect with regard to the interaction with CaMKII. CaMKII modulates memory retention in an N-methyl-D-aspartate receptor (NMDAR)-dependent manner in morphine-sensitized rats [17]. Reduced activations of CaMKII and cAMP response element-binding protein (CREB) were observed in the morphine self-administered rat NAc following treatment with a TRPV1 antagonist, which is from the transient receptor potential (TRP) cation channel family [18]. The autophosphorylation level of CaMKII in the limbic forebrain was increased in mice administered with morphine, and this upregulation was blocked by the intracerebroventricular infusion of KN-93 [19]. The CaMKII signal is fully involved in the effect of cocaine- and amphetamine-regulated transcript (CART) peptide, which is a neuropeptide associated with brain reward circuits, in the context of cocaine reward [20]. In the NAc, CaMKIIα contributes to the psychomotor effects induced by cocaine [21], reinstatement of cocaine-seeking behaviors [22], and cocaine-induced CPP behaviors [23][24]. CaMKIIα expressed in other brain regions such as the prefrontal cortex (PFC) and amygdala is also correlated with cocaine-induced CPP and cue-induced cocaine-seeking behaviors [23][25]. Furthermore, CaMKIIα autophosphorylation levels were significantly elevated in the PFC, hippocampus, and ventral striatum in rats following ketamine self-administration [26]. Here, we introduce some evidence suggesting that the activities of CaMKII (Table 1) are fully involved in certain brain regions located in the brain reward circuitry. Exposure to the stimulant drugs does not cause addiction immediately; it involves various neuronal adaptations that develop over time.
Table 1. Evidence of CaMKII activity fully involved in the brain regions of circuitry with various drugs of abuse.
| Drug | Region | CaMKII Activity |
|---|---|---|
| Nicotine | Nucleus accumbens | Phosphorylation level increased [12] |
| Hippocampal CA1 | Phosphorylation level increased [12] | |
| Ventral tegmental area | Activity increased [13] | |
| Morphine | Nucleus accumbens shell | Activity is crucial for morphine-seeking [27] |
| Hippocampus | CaMKIIα level increased in synaptosomes [28] | |
| Prefrontal cortex | Phosphorylation level increased [29] | |
| Cocaine | Nucleus accumbens shell | Activity is crucial for cocaine-seeking [21][22] |
| Hippocampal dentate gyrus | CaMKIIα phosphorylation mediates neuronal activation [28] | |
| Medial prefrontal cortex | CaMKIIα phosphorylation level increased after cocaine withdrawal [25] | |
| Amphetamine | Nucleus accumbens shell | Activity is crucial for amphetamine-induced DA release [30] |
| Hippocampus | Activity is crucial for amphetamine-induced CPP [31] | |
| Striatum | Phosphorylation level increased [32] |
These stimulant drugs have been reported to activate and alter the reward circuitry of the brain, and affect synaptic plasticity, learning acquisition, and glutamatergic inputs into the brain neuronal circuit [33][34] (Figure 1). The brain reward circuitry underlying the addiction process is complicated, which mainly includes DA release from DAergic neurons in the VTA of the midbrain and projections to the NAc and PFC region. CaMKIIα autophosphorylation levels were significantly elevated in the PFC of morphine-administered mice [29]. VTA plays a critical role in the initial stimulant drug exposure and causes long-term adaptation in DAergic neurons in the projection regions [35]. Stimulant drug exposure increases DA signaling in the NAc and functionally regulates glutamatergic excitatory projections to the NAc medium spiny neurons (MSNs). Addictive drugs trigger and modulate synaptic plasticity in the brain reward circuitry involved in addiction. Some studies have suggested that alterations in synaptic plasticity at glutamatergic inputs from the cortex to the NAc may be the underlying mechanism in stimulant drug addiction [33][36]. CaMKII is critically associated with the NAc shell DA and glutamatergic inputs involved in synaptic plasticity [22].
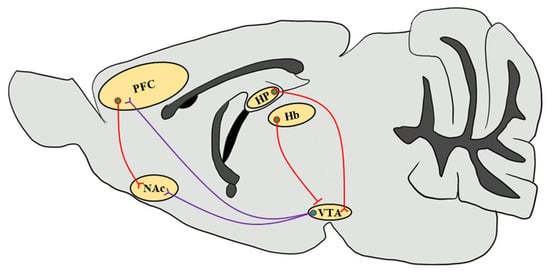
Figure 1. Simplified schematic representation of the mouse brain reward circuitry. The scheme of the mesolimbic DAergic projections (purple) of the mouse brain sagittal section emphasizes the predominant afferents that originate in the VTA and input into the NAc as well as the PFC, which are modulated by stimulant drug exposure through CaMKII functions. Glutamatergic inputs (red) that regulate neuronal circuits in response to drug abuse from the PFC to the NAc and from the HP to the VTA through the subiculum are highlighted [37]. Since CaMKIIβ regulates synaptic activity in the Hb in the context of depressive behaviors, it increases glutamatergic inputs onto VTA GABAergic interneurons to inhibit VTA DAergic neurons, thereby decreasing the DA release in the NAc [38][39]. VTA, ventral tegmental area; NAc, nucleus accumbens; PFC, prefrontal cortex; HP, hippocampus; Hb, habenula.
3. ERK in Nicotine and Other Psychostimulant-Induced Addiction
ERK is a serine-threonine protein kinase and a member of the mitogen-activated protein kinase (MAPK) family. It has been reported to play essential roles in cellular proliferation, differentiation, neuronal survival, and synaptic plasticity [40][41]. ERK has two isoforms, ERK1 (p44 MAPK) and ERK2, which are both expressed throughout the mesolimbic system in the mouse brain, including the regions such as the PFC, NAc, amygdala, and VTA [42]. ERK is recruited for signaling transfer in response to extracellular stimulations and the initiated signal cascade. BDNF activates receptor tyrosine kinases (RTKs), which are correlated with adapter protein recruitment such as Shc, and causes the activation of GTPase Ras, serine-threonine kinase Raf, and MAPK-ERK kinase (MEK), which in turn, phosphorylate and activate ERK. Moreover, Ras is also activated through L-type calcium channels and NMDAR. The Ca2+ influx evoked by the electrical and pharmacological activation of these channels and receptors plays a critical role in the signal cascade. Furthermore, dopamine D1 receptors (D1Rs) could also elevate Ca2+ influx and lead to the activation of protein kinase A (PKA) and Raf, which further causes ERK phosphorylation [43]. The phosphorylation of ERK leads to the activation of downstream targets such as Elk-1 transcription factor [44], and CREB through mitogen- and stress-stimulated kinase 1 (MSK1) and ribosomal S6 kinase (RSK) [45][46]. Since ERK is regulated by DAergic and glutamatergic signals, it has been suggested that ERK is involved in psychostimulant-induced addiction and plays a crucial role in the brain reward system and learning process [47]. Here, we provide a simplified schematic representation of the ERK signal cascade involved in psychostimulant-induced addiction (Figure 2).
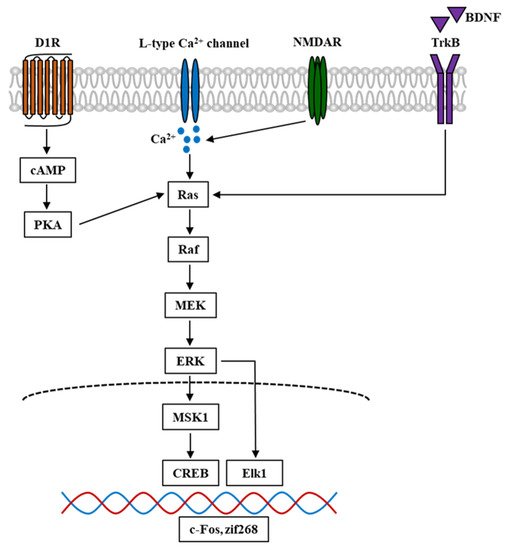
Figure 2. Simplified schematic diagram of ERK signal cascade in the mouse brain involved in psychostimulant-induced addiction. Nicotine and other stimulant drugs activate D1R and facilitate DA release in the DA terminals, leading to PKA and Raf activation. D1R activation causes L-type Ca2+ channels activation [48], contributing to Ca2+ influx and elevation of Ca2+ level, which leads to the activation of Raf. Raf is also activated by Ca2+ influx through NMDAR, which is mediated by the stimuli of Ras-GRF1 [49], and by BDNF through the binding and activation of TrkB. In turn, Raf activation leads to the phosphorylation and activation of MEK and ERK. The phosphorylation of ERK evokes the activation of downstream targets such as Elk1 and MSK1. The latter could activate and phosphorylate CREB. These transcriptional factors cause the transcription of IGEs such as c-Fos and zif268, which become the underlying mechanism of psychostimulant-induced addiction. D1R, dopamine D1 receptor; DA, dopamine; PKA, protein kinase A; NMDAR, N-methyl-D-aspartate receptor; Ras-GRF1, Ras protein-specific guanine-nucleotide releasing factor; BDNF, brain-derived neurotrophic factor; TrkB, tropomyosin receptor kinase B; MEK, MAPK-ERK kinase; MSK1, mitogen- and stress-stimulated kinase 1; CREB, cAMP response element-binding protein; IGEs, immediate early genes.
The activation of ERK is mediated by CaMKII in stimulant drug-induced addiction. Nicotine-induced elevation of ERK phosphorylation was found to be blocked in mouse primary cortical neurons following treatment with the CaMKII inhibitor KN-93 [50]. Treatment with KN-93 also significantly reduced nicotine-induced ERK phosphorylation levels in PC12h cells [51]. The calcium chelator BAPTA completely blocked nicotine-induced ERK phosphorylation, and L-type calcium channel blocker significantly decreased ERK phosphorylation levels following nicotine treatment, which suggests that Ca2+ is critical in the ERK phosphorylation process [50]. Moreover, the crosslinking and co-immunoprecipitation experiments proved that CaMKII forms a tight complex and interacts with NMDAR in living cells [52][53]. ERK phosphorylation induced by cocaine treatment is in accordance with the NMDAR function that NMDAR activation plays a role in neuronal adaptions involved in drug abuse [54]. Based on the evidence that Raf, the upstream target of ERK, is activated by Ca2+ influx through NMDAR, it is reasonable that CaMKII mediates and plays a critical role in ERK phosphorylation, which is involved in psychostimulant-induced addiction. Consistent with CaMKII, the activation of ERK is also involved in nicotine and other drugs of abuse. Some studies revealed that seven consecutive days of nicotine administration successfully elevated ERK phosphorylation levels in the rat NAc [55]. Subcutaneous administration of nicotine into adolescent rats caused a robust increase in ERK1/2 phosphorylation levels in the nucleus accumbens shell [56]. ERK1/2 phosphorylation levels were significantly upregulated following nicotine treatment in PC12h cells, while this elevation was inhibited by treatment with the MEK inhibitor, U0126 [51]. Furthermore, morphine induced significantly increased mice CPP behaviors, and ERK phosphorylation levels in the VTA were inhibited following treatment with the inhibitor, compound 511 [57]. Methamphetamine administration significantly increased the behavioral sensitization evaluated by the total distance in mice, and elevated ERK phosphorylation and ΔFosB levels in the mouse NAc [58]. Methamphetamine injection also elevated the expression of GluN2B, which in turn caused a significant increase in ERK, CREB phosphorylation, and BDNF levels [59]. Ethanol-induced enhancement of ethanol sensitization and ERK phosphorylation levels were observed in the mouse NAc shell, and these elevations were inhibited by pretreatment with MPEP, which is an mGluR5 antagonist [60].
References
- Forouzanfar, M.H.; Afshin, A.; Alexander, L.T.; Anderson, H.R.; Bhutta, Z.A.; Biryukov, S.; Brauer, M.; Burnett, R.; Cercy, K.; Charlson, F.J.; et al. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990–2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet 2016, 388, 1659–1724.
- Reitsma, M.B.; Fullman, N.; Ng, M.; Salama, J.S.; Abajobir, A.; Abate, K.H.; Abbafati, C.; Abera, S.F.; Abraham, B.; Abyu, G.Y.; et al. Smoking prevalence and attributable disease burden in 195 countries and territories, 1990–2015: A systematic analysis from the Global Burden of Disease Study 2015. Lancet 2017, 389, 1885–1906.
- Peto, R.; Boreham, J.; Lopez, A.; Thun, M.; Heath, C. Mortality from tobacco in developed countries: Indirect estimation from national vital statistics. Lancet 1992, 339, 1268–1278.
- Iribarren, C.; Tekawa, I.S.; Sidney, S.; Friedman, G.D. Effect of Cigar Smoking on the Risk of Cardiovascular Disease, Chronic Obstructive Pulmonary Disease, and Cancer in Men. N. Engl. J. Med. 1999, 340, 1773–1780.
- Rose, J.E.; Corrigall, W.A. Nicotine self-administration in animals and humans: Similarities and differences. Psychopharmacology 1997, 130, 28–40.
- Balfour, D.J.; Wright, A.E.; Benwell, M.E.; Birrell, C.E. The putative role of extra-synaptic mesolimbic dopamine in the neurobiology of nicotine dependence. Behav. Brain Res. 2000, 113, 73–83.
- Di Chiara, G. Role of dopamine in the behavioural actions of nicotine related to addiction. Eur. J. Pharmacol. 2000, 393, 295–314.
- Balfour, D.J.; Fagerström, K.O. Pharmacology of nicotine and its therapeutic use in smoking cessation and neurodegenerative disorders. Pharmacol. Ther. 1996, 72, 51–81.
- Shiffman, S.; Scharf, D.M.; Shadel, W.G.; Gwaltney, C.J.; Dang, Q.; Paton, S.M.; Clark, D.B. Analyzing milestones in smoking cessation: Illustration in a nicotine patch trial in adult smokers. J. Consult. Clin. Psychol. 2006, 74, 276–285.
- Lee, A.M.; Messing, R.O. Protein Kinases and Addiction. Ann. N. Y. Acad. Sci. 2008, 1141, 22–57.
- Li, C.-Y.; Mao, X.; Wei, L. Genes and (Common) Pathways Underlying Drug Addiction. PLoS Comput. Biol. 2008, 4, e2.
- Wilar, G.; Shinoda, Y.; Sasaoka, T.; Fukunaga, K. Crucial Role of Dopamine D2 Receptor Signaling in Nicotine-Induced Conditioned Place Preference. Mol. Neurobiol. 2019, 56, 7911–7928.
- Jackson, K.J.; Muldoon, P.P.; Walters, C.; Damaj, M.I. Neuronal calcium/calmodulin-dependent protein kinase II mediates nicotine reward in the conditioned place preference test in mice. Behav. Pharmacol. 2016, 27, 50–56.
- Risinger, F.; Oakes, R.A. Nicotine-induced conditioned place preference and conditioned place aversion in mice. Pharmacol. Biochem. Behav. 1995, 51, 457–461.
- Grabus, S.D.; Martin, B.R.; Brown, S.E.; Damaj, M.I. Nicotine place preference in the mouse: Influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology 2006, 184, 456–463.
- Le Foll, B.; Goldberg, S.R. Nicotine induces conditioned place preferences over a large range of doses in rats. Psychopharmacology 2005, 178, 481–492.
- Sadat-Shirazi, M.-S.; Ahmadian-Moghadam, H.; Khalifeh, S.; Zadeh-Tehrani, S.N.; Farahmandfar, M.; Zarrindast, M.-R. The role of calcium-calmodulin-dependent protein kinase II in modulation of spatial memory in morphine sensitized rats. Behav. Brain Res. 2019, 359, 298–303.
- Ma, S.X.; Kim, H.C.; Lee, S.Y.; Jang, C.G. TRPV1 modulates morphine self-administration via activation of the CaMKII-CREB pathway in the nucleus accumbens. Neurochem. Int. 2018, 121, 1–7.
- Narita, M.; Matsumura, Y.; Ozaki, S.; Ise, Y.; Yajima, Y.; Suzuki, T. Role of the calcium/calmodulin-dependent protein kinase ii (CaMKII) in the morphine-induced pharmacological effects in the mouse. Neuroscience 2004, 126, 415–421.
- Yu, C.; Zhou, X.; Fu, Q.; Peng, Q.; Oh, K.-W.; Hu, Z. A New Insight into the Role of CART in Cocaine Reward: Involvement of CaMKII and Inhibitory G-Protein Coupled Receptor Signaling. Front. Cell. Neurosci. 2017, 11, 244.
- Cates, H.M.; Thibault, M.; Pfau, M.; Heller, E.; Eagle, A.; Gajewski, P.; Bagot, R.; Colangelo, C.; Abbott, T.; Rudenko, G.; et al. Threonine 149 phosphorylation enhances DeltaFosB transcriptional activity to control psychomotor responses to cocaine. J. Neurosci. 2014, 34, 11461–11469.
- Anderson, S.M.; Famous, K.R.; Sadri-Vakili, G.; Kumaresan, V.; Schmidt, H.D.; Bass, C.E.; Terwilliger, E.F.; Cha, J.H.; Pierce, R.C. CaMKII: A biochemical bridge linking accumbens dopamine and glutamate systems in cocaine seeking. Nat. Neurosci. 2008, 11, 344–353.
- Kong, H.; Kuang, W.; Li, S.; Xu, M. Activation of dopamine D3 receptors inhibits reward-related learning induced by cocaine. Neuroscience 2011, 176, 152–161.
- Easton, A.C.; Lourdusamy, A.; Havranek, M.; Mizuno, K.; Solati, J.; Golub, Y.; Clarke, T.K.; Vallada, H.; Laranjeira, R.; Desrivières, S.; et al. alphaCaMKII controls the establishment of cocaine’s reinforcing effects in mice and humans. Transl. Psychiatry 2014, 4, e457.
- Caffino, L.; Cassina, C.; Giannotti, G.; Di Clemente, A.; Racagni, G.; Fumagalli, F.; Cervo, L.; Orrù, A.; Moro, F. Short-term abstinence from cocaine self-administration, but not passive cocaine infusion, elevates ?CaMKII autophosphorylation in the rat nucleus accumbens and medial prefrontal cortex. Int. J. Neuropsychopharmacol. 2013, 17, 323–329.
- Caffino, L.; Piva, A.; Mottarlini, F.; Di Chio, M.; Giannotti, G.; Chiamulera, C.; Fumagalli, F. Ketamine Self-Administration Elevates alphaCaMKII Autophosphorylation in Mood and Reward-Related Brain Regions in Rats. Mol. Neurobiol. 2018, 55, 5453–5461.
- Liu, Z.; Zhang, J.-J.; Liu, X.-D.; Yu, L.-C. Inhibition of CaMKII activity in the nucleus accumbens shell blocks the reinstatement of morphine-seeking behavior in rats. Neurosci. Lett. 2012, 518, 167–171.
- Schopf, I.; Easton, A.C.; Solati, J.; Golub, Y.; Kornhuber, J.; Giese, K.P.; Müller, C.P. alphaCaMKII autophosphorylation mediates neuronal activation in the hippocampal dentate gyrus after alcohol and cocaine in mice. Neurosci. Lett. 2015, 591, 65–68.
- Hu, X.; Huang, F.; Szymusiak, M.; Liu, Y.; Wang, Z.J. Curcumin attenuates opioid tolerance and dependence by inhibiting Ca2+/calmodulin-dependent protein kinase II α activity. J. Pharmacol. Exp. Ther. 2014, 352, 420–428.
- Pierce, R.C.; Kalivas, P.W. Repeated Cocaine Modifies the Mechanism by which Amphetamine Releases Dopamine. J. Neurosci. 1997, 17, 3254–3261.
- Tan, S.-E. Impairing the amphetamine conditioning in rats through the inhibition of hippocampal calcium/calmodulin-dependent protein kinase II activity. Neuropharmacology 2002, 42, 540–547.
- Choe, E.S.; Wang, J.Q. CaMKII regulates amphetamine-induced ERK1/2 phosphorylation in striatal neurons. Neuroreport 2002, 13, 1013–1016.
- Kauer, J.A.; Malenka, R.C. Synaptic plasticity and addiction. Nat. Rev. Neurosci. 2007, 8, 844–858.
- Robison, A.J.; Nestler, E.J. Transcriptional and epigenetic mechanisms of addiction. Nat. Rev. Neurosci. 2011, 12, 623–637.
- Kauer, J.A. Learning Mechanisms in Addiction: Synaptic Plasticity in the Ventral Tegmental Area as a Result of Exposure to Drugs of Abuse. Annu. Rev. Physiol. 2004, 66, 447–475.
- Kalivas, P.W. The glutamate homeostasis hypothesis of addiction. Nat. Rev. Neurosci. 2009, 10, 561–572.
- Vorel, S.R.; Liu, X.; Hayes, R.J.; Spector, J.A.; Gardner, E.L. Relapse to Cocaine-Seeking After Hippocampal Theta Burst Stimulation. Science 2001, 292, 1175–1178.
- Li, K.; Zhou, T.; Liao, L.; Yang, Z.; Wong, C.; Henn, F.; Malinow, R.; Yates, J.R.; Hu, H. betaCaMKII in lateral habenula mediates core symptoms of depression. Science 2013, 341, 1016–1020.
- Ji, H.; Shepard, P.D. Lateral habenula stimulation inhibits rat midbrain dopamine neurons through a GABA(A) receptor-mediated mechanism. J. Neurosci. 2007, 27, 6923–6930.
- Kyosseva, S.V. Mitogen-Activated Protein Kinase Signaling. Int. Rev. Neurobiol. 2004, 59, 201–220.
- Hetman, M.; Gozdz, A. Role of extracellular signal regulated kinases 1 and 2 in neuronal survival. JBIC J. Biol. Inorg. Chem. 2004, 271, 2050–2055.
- Lein, E.S.; Hawrylycz, M.J.; Ao, N.; Ayres, M.; Bensinger, A.; Bernard, A.; Boe, A.F.; Boguski, M.S.; Brockway, K.S.; Byrnes, E.J.; et al. Genome-wide atlas of gene expression in the adult mouse brain. Nat. Cell Biol. 2006, 445, 168–176.
- Lu, L.; Koya, E.; Zhai, H.; Hope, B.T.; Shaham, Y. Role of ERK in cocaine addiction. Trends Neurosci. 2006, 29, 695–703.
- Valjent, E.; Corvol, J.-C.; Pagès, C.; Besson, M.-J.; Maldonado, R.; Caboche, J. Involvement of the Extracellular Signal-Regulated Kinase Cascade for Cocaine-Rewarding Properties. J. Neurosci. 2000, 20, 8701–8709.
- Mattson, B.J.; Bossert, J.M.; Simmons, D.E.; Nozaki, N.; Nagarkar, D.; Kreuter, J.D.; Hope, B.T. Cocaine-induced CREB phosphorylation in nucleus accumbens of cocaine-sensitized rats is enabled by enhanced activation of extracellular signal-related kinase, but not protein kinase A. J. Neurochem. 2005, 95, 1481–1494.
- Brami-Cherrier, K.; Valjent, E.; Hervé, D.; Darragh, J.; Corvol, J.-C.; Pagès, C.; Simon, A.J.; Girault, J.-A.; Caboche, J. Parsing Molecular and Behavioral Effects of Cocaine in Mitogen- and Stress-Activated Protein Kinase-1-Deficient Mice. J. Neurosci. 2005, 25, 11444–11454.
- Girault, J.A.; Valjent, E.; Caboche, J.; Herve, D. ERK2: A logical AND gate critical for drug-induced plasticity? Curr. Opin. Pharmacol. 2007, 7, 77–85.
- Surmeier, D.; Bargas, J.; Hemmings, H.C., Jr.; Nairn, A.C.; Greengard, P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron 1995, 14, 385–397.
- Farnsworth, C.L.; Freshney, N.W.; Rosen, L.B.; Ghosh, A.; Greenberg, M.E.; Feig, L.A. Calcium activation of Ras mediated by neuronal exchange factor Ras-GRF. Nat. Cell Biol. 1995, 376, 524–527.
- Steiner, R.C.; Heath, C.J.; Picciotto, M.R. Nicotine-induced phosphorylation of ERK in mouse primary cortical neurons: Evidence for involvement of glutamatergic signaling and CaMKII. J. Neurochem. 2007, 103, 666–678.
- Nakayama, H.; Numakawa, T.; Ikeuchi, T.; Hatanaka, H. Nicotine-induced phosphorylation of extracellular signal-regulated protein kinase and CREB in PC12h cells. J. Neurochem. 2008, 79, 489–498.
- Gardoni, F.; Caputi, A.; Cimino, M.; Pastorino, L.; Cattabeni, F.; Di Luca, M. Calcium/calmodulin-dependent protein kinase II is associated with NR2A/B subunits of NMDA receptor in postsynaptic densities. J. Neurochem. 2002, 71, 1733–1741.
- Leonard, A.S.; Lim, I.A.; Hemsworth, D.E.; Horne, M.C.; Hell, J.W. Calcium/calmodulin-dependent protein kinase II is associated with the N-methyl-D-aspartate receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 3239–3244.
- Delint-Ramirez, I.; Segev, A.; Pavuluri, A.; Self, D.W.; Kourrich, S. Cocaine-Induced Synaptic Redistribution of NMDARs in Striatal Neurons Alters NMDAR-Dependent Signal Transduction. Front. Neurosci. 2020, 14, 698.
- Yang, J.H.; Sohn, S.; Kim, S.; Kim, J.; Oh, J.H.; Ryu, I.S.; Go, B.S.; Choe, E.S. Repeated nicotine exposure increases the intracellular interaction between ERK-mGluR5 in the nucleus accumbens more in adult than adolescent rats. Addict. Biol. 2021, 26, e12913.
- Hudson, R.; Green, M.; Wright, D.J.; Renard, J.; Jobson, C.E.; Jung, T.; Rushlow, W.; Laviolette, S.R. Adolescent nicotine induces depressive and anxiogenic effects through ERK 1-2 and Akt-GSK-3 pathways and neuronal dysregulation in the nucleus accumbens. Addict. Biol. 2021, 26, e12891.
- Zhang, H.; Wang, Q.; Sun, Q.; Qin, F.; Nie, D.; Li, Q.; Gu, Y.; Jiang, Y.; Lu, S.; Lu, Z. Effects of Compound 511 on BDNF-TrkB Signaling in the Mice Ventral Tegmental Area in Morphine-Induced Conditioned Place Preference. Cell. Mol. Neurobiol. 2020.
- Wen, D.; Hui, R.; Liu, Y.; Luo, Y.; Wang, J.; Shen, X.; Xie, B.; Yu, F.; Cong, B.; Ma, C. Molecular hydrogen attenuates methamphetamine-induced behavioral sensitization and activation of ERK-DeltaFosB signaling in the mouse nucleus accumbens. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 97, 109781.
- Kim, B.; Jha, S.; Seo, J.H.; Jeong, C.-H.; Lee, S.; Lee, S.; Seo, Y.H.; Park, B. MeBib Suppressed Methamphetamine Self-Administration Response via Inhibition of BDNF/ERK/CREB Signal Pathway in the Hippocampus. Biomol. Ther. 2020, 28, 519–526.
- Stevenson, R.A.; Hoffman, J.L.; Maldonado-Devincci, A.M.; Faccidomo, S.; Hodge, C.W. MGluR5 activity is required for the induction of ethanol behavioral sensitization and associated changes in ERK MAP kinase phosphorylation in the nucleus accumbens shell and lateral habenula. Behav. Brain Res. 2019, 367, 19–27.
More
Information
Subjects:
Neurosciences
Contributor
MDPI registered users' name will be linked to their SciProfiles pages. To register with us, please refer to https://encyclopedia.pub/register
:
View Times:
986
Revisions:
3 times
(View History)
Update Date:
03 Jun 2021
Table of Contents



Notice
You are not a member of the advisory board for this topic. If you want to update advisory board member profile, please contact office@encyclopedia.pub.
OK
Confirm
Only members of the Encyclopedia advisory board for this topic are allowed to note entries. Would you like to become an advisory board member of the Encyclopedia?
Yes
No
${ textCharacter }/${ maxCharacter }
Submit
Cancel
Back
Comments
${ item }
|
${ item.createdUser.fullName }
${ item.createdAt }
${ item.vote }
${ item.reply }
Delete
${ reply.createdUser.fullName }
${ reply.createdAt }
${ reply.vote }
Delete
There is no reply to this comment~
${ item.replyTextCharacter }/${ item.replyMaxCharacter }
Submit
Cancel
More
No more~
There is no comment~
${ textCharacter }/${ maxCharacter }
Submit
Cancel
${ selectedItem.replyTextCharacter }/${ selectedItem.replyMaxCharacter }
Submit
Cancel
Confirm
Are you sure to Delete?
Yes
No