1000/1000
Hot
Most Recent
 +1 point
+1 point
Epstein–Barr virus (EBV) is a linear, double-stranded DNA gamma herpes virus which affects more than 90% of the worldwide population. Clinically, EBV primo-infection is usually silent, but symptomatic forms may arise in children and young adults, leading to infectious mononucleosis, where B lymphocytes are highly infected. In most EBV-infected individuals, the virus establishes lifelong latent infection with no specific clinical manifestation. However, EBV primary infections, as well as secondary reactivations, may cause various EBV-associated lymphoproliferative disorders (LPD) including hematologic malignancies, especially within immunocompromised hosts. While most of EBV-associated LPD involve B-cells, EBV-associated T/NK LPD are also reported, mostly appearing in Asia and Latin America and representing a spectrum of reactive, indolent and malignant disorders.
Epstein–Barr virus (EBV) is a linear, double-stranded DNA gamma herpes virus which affects more than 90% of the worldwide population [1]. EBV primo-infection occurs through oral transmission in oropharyngeal epithelial cells, where the virus undergoes its first lytic replication and expresses its viral DNA genome, resulting in the production of new viral particles. Then, EBV affects naïve B-cells and establishes latent infection, characterized by the persistence of episomal viral genomes in the nuclei of memory B-cells. These cells can therefore further circulate in peripheral blood and constitute the main persistent EBV reservoir. There are three types of latency programs, based on the expression or co-expression of EBV latency molecular markers, including six nuclear antigens (EBNA1, EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-LP), three latent membrane proteins (LMP1, LMP2a and LMP2B) and two short non-coding RNAs (EBER1 and EBER2). Expression of the full set of EBV-encoded proteins, referred to as type III latency program, is activated in infected naïve B-cells following EBV primo-infection. In order to escape immune surveillance, and to allow infected cells to survive, there is a restriction in the latent gene transcription machinery with a lack of EBNA2 expression (type II latency). Finally, the type I latency program is limited to the expression of EBNA1 and EBERs and is involved in the maintenance and replication of the episomal viral genome [2]. EBV latent antigens are able to impact both the growth and transformation of B-cells, which may lead to the development of subsequent lymphomas. Cells with restricted EBV gene expression (latency I and II) present no autonomous growth potential, thus malignant transformation of such cells requires additional factors (genetic/cytogenetic changes, cytokines or cellular interactions) [2]. Two genetically different EBV forms (EBV type 1 and 2) are distinguished by allelic polymorphisms in the latent-cycle genes encoding the nuclear antigens 2 and 3 (EBNA2 and EBNA3, respectively). Both types are detected worldwide, type 1 being the most prevalent, except in central Africa, Papua New Guinea and Alaska, where type 2 is more common [3].
Hemophagocytic lymphohistiocytosis (HLH) is a fulminant life-threatening inflammatory disease due to uncontrolled hyperactivation of the immune system. Its diagnosis is based on the presence of 5 out of 8 of the following HLH criteria: fever, splenomegaly, cytopenias, hypertriglyceridemia or hypofribrinogenemia, hemophagocytosis, low or absent NK-cell activity, hyperferritinemia, and high soluble CD25 levels [4]. However, these symptoms and biological abnormalities are not specific to HLH, and some of them may occur during any trivial primary EBV infection. Timing and severity of these signs help make the distinction.
Two types of HLH are recognized: a primary (i.e., familial) form related to various genetic alterations, and a secondary one [5][6][7]. Most of the genetic alterations described in primary HLH impair NK- or T-cell functions—PRF1, UNC13D, STX11, STXBP2, and SH2D1A mutations causing familial HLH2, HLH3, HLH4, HLH5, and X-linked lymphoproliferative disease type 1 (XLP1), respectively. On the contrary, other less common primary HLH such as the X-linked lymphoproliferative disease type 2 (XLP2) present normal cytotoxicity and are due to dysregulated inflammasome or TNFR responses [8][9]. In the absence of any genetic predisposition, secondary HLH is commonly triggered by infections, malignancies or autoimmune disorders, EBV being the most common infection associated with it [5][7][10].
EBV–positive HLH is rare and mainly affects children and adolescents in Asia [11]. Histological analyses show lymphohistiocytic infiltrates with hemophagocytosis and EBV-infected cells, which are small and show slight or no atypia (Figure 1).
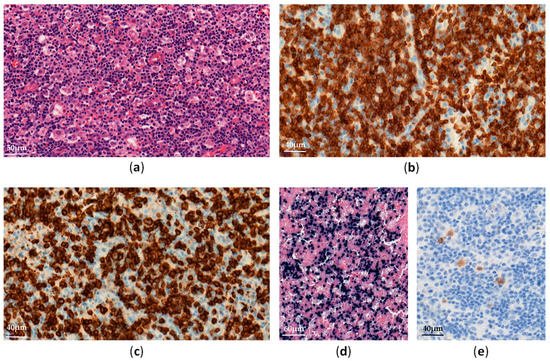
Figure 1. EBV-positive hemophagocytic lymphohistiocytosis in a lymph node: (a) Diffuse lymphohistiocytic infiltrate with hemophagocytosis (H&E ×300); (b–e) Atypical lymphoid cells are positive for CD3 (b, ×400), CD8 (c, ×400), EBV-encoded small RNA (EBER) (d, ×200) and LMP1 (e, ×400).
EBV-infected cells can be observed in many organs (e.g., lymph nodes, liver, spleen, bone marrow). These cells often have a cytotoxic CD8+ T-cell origin and less frequently an NK-cell phenotype [12]. TCR monoclonal rearrangements may be found in T-cell HLH, due to clonal proliferation of EBV-infected T-cells, but do not correlate with clinical outcomes [13].
Survival for patients with EBV-associated HLH has greatly improved when treated promptly in accordance with the HLH-2004 protocol [4]. However, some patients’ conditions evolve into other EBV-positive T/NK LPD requiring more intensive therapies. In addition, EBV-HLH may also occur at the same time as systemic EBV-positive T/NK-cell lymphoma of childhood.
Systemic chronic active EBV infection (CAEBV) is defined as a chronic systemic illness related to EBV infection. It is characterized by persistent clinical symptoms for more than 3 months, including fever, hepatosplenomagly and lymphadenopathy, without any evidence of underlying immunodeficiency [14]. Most cases have been reported in Asia and Latin America, involving predominantly T- or NK-cells, but EBV-infected B-cells have also been reported in CAEBV patients from the USA [15].
Histopathological analyses of affected tissues are not specific to CAEBV and usually show non-destructive reactive inflammatory lesions [14]. The diagnosis is made through the detection of elevated EBV DNA levels in peripheral blood and EBV-positive lymphocytes in tissues, showing a type II latency pattern (Figure 2). Monitoring EBV-DNA loads in peripheral blood is also useful for assessing the treatment response and appears to have prognostic value [16].
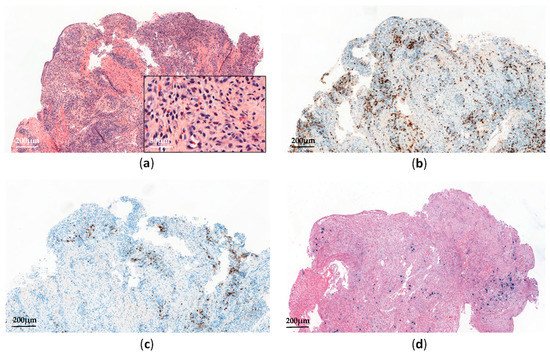
Figure 2. Systemic chronic active EBV infection involving the gastrointestinal tract: (a) Esophagus biopsy shows a polymorphic inflammatory infiltrate with a predominance of non-atypical small lymphocytes (H&E, ×60 and ×300); (b) The lymphoid infiltrate contains a predominance of CD3+ T-cells (×60); (c) Anti-CD20 immunostaining shows scattered reactive B-cells (×60); (d) EBV-encoded small RNA (EBER) in situ hybridization shows scattered positive cells (×60).
In Asia, most CAEBV are of T-cell type (60%), being mostly CD4+ rather than CD8+ [17][18]. However, some double positivity (CD4+ and CD8+) and γδ phenotypes have also been reported [15]. Chromosomal aberrations, increasing during the clinical course, were detected in some cases, and monoclonal EBV-infected T-cells have been reported in 50% of patients [17]. Although no genetic defects have been identified yet, some CAEBV families and racial susceptibilities have been described, suggesting genetic predispositions to EBV-mediated immune dysregulations [19][20]. In fact, HLA 26 and 52 loci—frequently seen in Asia and Mexico, respectively—have recently been reported with a higher risk of EBV-positive T/NK LPD [21]. The CAEBV clinical course ranges from indolent presentations (with episodic symptoms and asymptomatic periods) to fulminant presentations leading to death in a few weeks, in the case of no therapeutic intervention [21]. Patients with T-cell type CAEBV present worse outcomes than those with NK-cell type [18][22]. However, some NK-cell CAEBV may also evolve into aggressive NK-cell leukemia (ANKL) or extra-nodal NK/T-cell lymphoma, nasal type (ENKTL) [22]. In a comprehensive review of CAEBV, Arai described recent evidence suggesting the involvement of NF-κB and JAK/STAT pathways in the development of both inflammatory and neoplastic CAEBV aspects [23]. Moreover, a recent whole-exome sequencing analysis performed on 83 CAEBV patients has identified recurrent somatic mutations involving DDX3X, KMT2D, BCOR/BCORL1, KDM6A, and TET2 genes, with at least one somatic mutation detected in 58% of cases [24]. Interestingly, DDX3X mutations—known to be associated with hematological malignancies such as Burkitt lymphoma and ENKTL [25][26]—have been reported in serial lymphomas arising from previous CAEBV [21]. This suggests that in such cases, the acquisition of DDX3X mutations among others may trigger lymphomagenesis. More recently, Okuno et al. have also reported that EBV genomes in CAEBV patients harbored frequent intragenic deletions that frequently occurred in the BamHI rightward transcript microRNA clusters and several genes required for producing viral particles [27]. Such deletions are expected to reactivate the lytic cycle (by upregulating the expression of two immediate early genes, namely BZLF1 and BRLF) and to preclude viral production and cell lysis [27]. Finally, in a recent review article on CAEBV, the authors detailed the potential role of host genetic factors in the pathogenesis of CAEBV. While CAEBV develops in immunocompetent hosts by definition, some patients appear to have minor defects in cellular immunity that may impair immunosurveillance on EBV-infected T/NK-cells [28][29][30].
According to the clinical course and histological data, Ohshima et al. have proposed a classification of systemic CAEBV forms as follows: Category A1 (polyclonal and polymorphic LPD), Category A2 (monoclonal and polymorphic LPD), and Category A3 (monoclonal and monomorphic LPD) [31]. The fourth one, called Category B (monoclonal and monomorphic LPD with a fulminant course), has been considered to be a systemic EBV-positive T-cell lymphoma of childhood in the 2017 World Health Organization (WHO) classification (see below) [14]. Importantly, monoclonality in the proliferation predicts no increase in the mortality rate and should not be diagnosed as a lymphoma [22][31].
Hydroavacciniforme-like lymphoproliferative disorder (HV-LPD) is a rare cutaneous form of CAEBV characterized by blistering photodermatoses with vacciniform scars [32][33][34]. It mostly affects children in Asia and Latin America, but adult and elderly cases have also been reported [34][35]. HV-LPD has a broad range of clinical presentations ranging from indolent cutaneous forms to the severe systemic form [14][34]. Classic HV-LPD exhibits lesions typically limited to photoexposed skin, displaying favorable outcomes as it spontaneously regresses in adulthood. However, severe forms of HV-LPD also exist, characterized by extensive and ulcerative skin lesions with systemic symptoms [17][33][34]. Such HV-LPD may eventually evolve into other EBV-positive T/NK LPD, such as systemic CAEBV or systemic EBV-positive T-cell lymphoma, ENKTL, or ANKL [34]. Recent data has suggested that white patients with HV-LPD are less likely to progress into systemic diseases and show a much better prognosis than non-white patients [36]. Skin biopsies show inflammatory infiltrates with necrosis, ulceration, and angiocentricity. EBV-infected cells with type II latency are found in variable proportions and are small or medium-sized cells with cytotoxic CD8+ T-cell (70%) rather than NK-cell phenotypes [33][34].
HV-LPD pathogenesis remains unclear, but like in other EBV-positive T/NK LPD, geographic and racial distributions suggest some genetic predispositions. TCR gene monoclonal rearrangements are found in almost all T-cell type HV-LPD, with no prognosis impact [33][37]. Genomic explorations of HV-LPD have rarely been carried out. A recent whole-exome sequencing analysis performed in five Chinese patients with HV-LPD found five potentially driver mutations involving STAT3, IKBKB, ELF3, CHD7, and KMT2D genes [38], but these findings need to be confirmed for larger cohorts.
Severe mosquito bite allergy (SMBA) is another cutaneous manifestation of CAEBV, mostly reported in Asia and Mexico [17][39]. SMBA mainly affects children and adolescents and is characterized by localized skin lesions (erythema, bullae, and ulceration) with variable systemic symptoms (fever, lymphadenopathy, and liver dysfunction) appearing some hours after a mosquito bite. Patients present increased serum IgE titers and high blood EBV DNA loads with NK-cell lymphocytosis [14].
Histologically, skin biopsies show similar lesions to HV-LPD, with more extensive local necrosis and angiodestruction associated with a polymorphic infiltrate rich in eosinophils and histiocytes, admixed with small lymphocytes and larger atypical cells. EBV-infected cells (type II latency) are found in fewer proportions than in HV-LPD and usually have an NK-cell phenotype (CD3ε+, CD56+) [14]. Chromosomal alterations have rarely been identified [17], but full molecular studies on SMBA are lacking in the literature. Finally, SMBA may worsen due to HLH or evolve into other EBV-positive T/NK LPD, such as HV-LPD, systemic CAEBV, or even NK/T-cell lymphoma or ANKL [17].
Systemic EBV-positive T-cell lymphoma of childhood (STCLC) is a fulminant systemic T-cell lymphoma that usually occurs in Asian children and young adults, shortly after EBV primo-infection or sometimes following systemic CAEBV (especially monoclonal ones) [14][20][40]. STCLC patients present systemic symptoms with HLH almost always rapidly triggering multi-organ failure and death. Biopsies of affected tissues show proliferation of bland small or medium-sized EBV+ T-cells, sometimes admixed with large and atypical cells [14]. Primary STCLC usually have a cytotoxic CD8+ phenotype, while STCLC that happens after CAEBV are mostly CD4+ [40]. The EBV latency program has not been clearly established yet, and LMP1 is usually negative by immunohistochemistry.
Most of the cases show monoclonal TCR rearrangements [14][41]. Moreover, chromosomal aberrations have been reported in some cases and turned out to be associated with the poorest outcomes [42]. As STCLC have many clinicopatholgical features overlapping with EBV-associated HLH, the identification of karyotypic abnormalities should therefore be helpful to distinguish these two entities. In a recent retrospective study of targeted NGS in a cohort of 169 EBV T/NK-LPD, including 34 STCLC, mutations were detected in 88.2% of STCLC cases, with KMT2D being the most frequently mutated gene (17.6%), followed by MFHAS1 (14.7%), STAT3 (14.7%), EP300 (11.8%), ITPKB (8.8%), DDX3X (8.8%), NOTCH1 (8.8%), and NOTCH2 (8.8%) [43].





