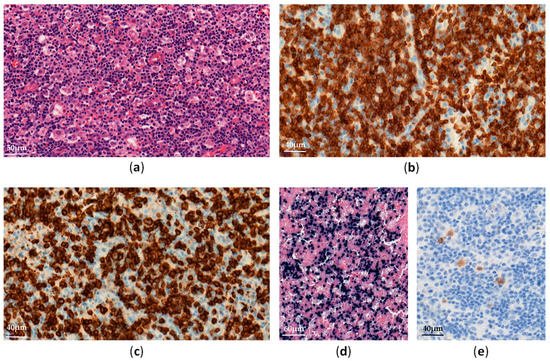
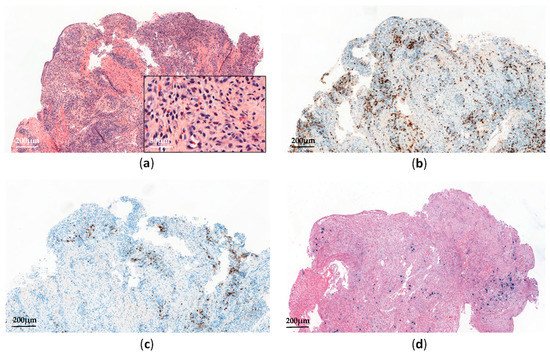
Systemic chronic active EBV infection (CAEBV) is defined as a chronic systemic illness related to EBV infection. It is characterized by persistent clinical symptoms for more than 3 months, including fever, hepatosplenomagly and lymphadenopathy, without any evidence of underlying immunodeficiency [
19]. Most cases have been reported in Asia and Latin America, involving predominantly T- or NK-cells, but EBV-infected B-cells have also been reported in CAEBV patients from the USA [
20].
In Asia, most CAEBV are of T-cell type (60%), being mostly CD4+ rather than CD8+ [
4,
22]. However, some double positivity (CD4+ and CD8+) and γδ phenotypes have also been reported [
20]. Chromosomal aberrations, increasing during the clinical course, were detected in some cases, and monoclonal EBV-infected T-cells have been reported in 50% of patients [
4]. Although no genetic defects have been identified yet, some CAEBV families and racial susceptibilities have been described, suggesting genetic predispositions to EBV-mediated immune dysregulations [
23,
24]. In fact, HLA 26 and 52 loci—frequently seen in Asia and Mexico, respectively—have recently been reported with a higher risk of EBV-positive T/NK LPD [
25]. The CAEBV clinical course ranges from indolent presentations (with episodic symptoms and asymptomatic periods) to fulminant presentations leading to death in a few weeks, in the case of no therapeutic intervention [
25]. Patients with T-cell type CAEBV present worse outcomes than those with NK-cell type [
22,
26]. However, some NK-cell CAEBV may also evolve into aggressive NK-cell leukemia (ANKL) or extra-nodal NK/T-cell lymphoma, nasal type (ENKTL) [
26]. In a comprehensive review of CAEBV, Arai described recent evidence suggesting the involvement of NF-κB and JAK/STAT pathways in the development of both inflammatory and neoplastic CAEBV aspects [
27]. Moreover, a recent whole-exome sequencing analysis performed on 83 CAEBV patients has identified recurrent somatic mutations involving
DDX3X,
KMT2D,
BCOR/BCORL1,
KDM6A, and
TET2 genes, with at least one somatic mutation detected in 58% of cases [
28]. Interestingly,
DDX3X mutations—known to be associated with hematological malignancies such as Burkitt lymphoma and ENKTL [
29,
30]—have been reported in serial lymphomas arising from previous CAEBV [
25]. This suggests that in such cases, the acquisition of
DDX3X mutations among others may trigger lymphomagenesis. More recently, Okuno et al. have also reported that EBV genomes in CAEBV patients harbored frequent intragenic deletions that frequently occurred in the
BamHI rightward transcript microRNA clusters and several genes required for producing viral particles [
31]. Such deletions are expected to reactivate the lytic cycle (by upregulating the expression of two immediate early genes, namely
BZLF1 and
BRLF) and to preclude viral production and cell lysis [
31]. Finally, in a recent review article on CAEBV, the authors detailed the potential role of host genetic factors in the pathogenesis of CAEBV. While CAEBV develops in immunocompetent hosts by definition, some patients appear to have minor defects in cellular immunity that may impair immunosurveillance on EBV-infected T/NK-cells [
32,
33,
34].
According to the clinical course and histological data, Ohshima et al. have proposed a classification of systemic CAEBV forms as follows: Category A1 (polyclonal and polymorphic LPD), Category A2 (monoclonal and polymorphic LPD), and Category A3 (monoclonal and monomorphic LPD) [
35]. The fourth one, called Category B (monoclonal and monomorphic LPD with a fulminant course), has been considered to be a systemic EBV-positive T-cell lymphoma of childhood in the 2017 World Health Organization (WHO) classification (see below) [
19]. Importantly, monoclonality in the proliferation predicts no increase in the mortality rate and should not be diagnosed as a lymphoma [
26,
35].