 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Joanna Wierońska | + 1555 word(s) | 1555 | 2021-08-05 11:30:34 | | | |
| 2 | Amina Yu | Meta information modification | 1555 | 2021-08-19 03:56:57 | | |
Video Upload Options
The review summarizes the roles of NO•-dependent pathways in the early and late aftermath of stroke and treatments based on the stimulation or inhibition of particular NO• synthases and the stabilization of HIF-1α activity.
1. Brain Ischemic Stroke and the Role of NO Its Pathology
Among the variety of factors that contribute to the cellular events leading to ischemic neuronal death, the fundamental factors are N-methyl-D-aspartate receptor (NMDA)-induced excitotoxicity and NO • -dependent pathways which are functionally linked.
In the physiological state, glutamate activates the synaptic pool of NMDA receptors
and is rapidly taken up from the synaptic space by presynaptic mechanisms and astrocytes. Opening of NMDA ion channels and subsequent Ca2+ influx into the neurons activates constitutive nitric oxide synthases (cNOS) to produce nitric oxide (NO). Under pathological conditions, due to the “uncoupling” of NOS activity from electron donation by NADPH and/or reduced availability of L-arginine or BH4, electrons are transferred from NADPH to flavins via the reductase domain, forming superoxide (O2 •−), and other reactive oxygen species (ROS). NO• reacts with O2 •− to form reactive nitrogen species (RNS) including the highly toxic peroxynitrite (ONOO−), leading to the production of other secondary components of nitroxidative stress, such as NO2+, NO2 and OH•, which initiate a cascade of redox reactions. In contrast to cNOS, the activity of inducible NO• synthase (iNOS) is Ca2+-independent. The enzyme is activated mostly in glial cells or leukocytes by certain cytokines, such as interferon-γ, tumor necrosis factor-α or interleukin-1β, in response to inflammation iNOS produces much greater amounts of NO• than both eNOS and nNOS combined. Therefore, iNOS is often referred to as the “pathological” form of NO• synthase, as it may promote the production of ONOO− and subsequently highly reactive hydroxyl radicals.
he distribution and concentration of NO• in brain tissue change after ischemic
episode and differs both temporally and spatially postinjury. Time-dependent changes induced by ischemia are related in particular to the activity of individual NOS isoforms, and in general, NO• produced by nNOS or iNOS plays detrimental role; while eNOS is neuroprotective. The expression of nNOS increases rapidly, in parallel with membrane depolarization and Ca2+ elevation in the cells, while massive production of iNOS is observed several hours after ischemic episode.
Neurons are particularly sensitive to stress caused by ROS/RNS overproduction because of the relatively low levels of antioxidants compared with other cells. Physiologically, basal ROS/RNS production in neurons is constitutively generated by mitochondrial metabolism, which is higher than that in other cells due to the necessity to maintain neuronal circuit activity and synaptic transmission. Excessive nNOS activation under hyperactivation of NMDA receptors (i.e due to ischemic episode) sresults in massive and uncontrolled generation of NO • followed by the downstream formation of ROS/RNS, resulting in neuroinflammation and nitro oxidative burst. The prolonged destabilization of nNOS contributes to neurotoxicity and the potentiation of ischemic damage ( Figure 1).
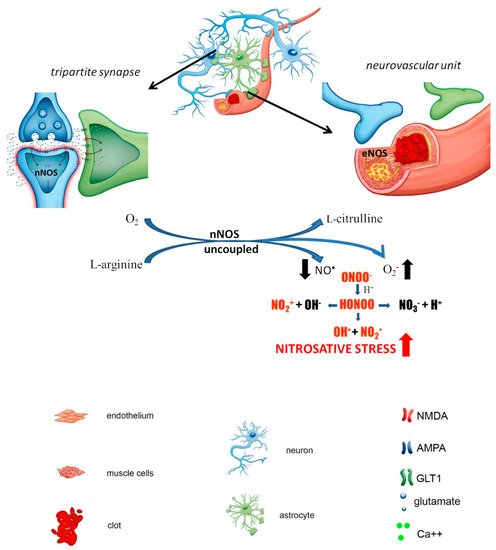
ischemia. Astrocytes play critical role BBB integrity and the maintenance of extracellular ion homeostasis by buffering excitatory transmitters released by neurons and producing trophic factors that support neuronal growth and survival. In response to pathological situations in surrounding tissue such as ischemic injury astrocytes undergo morphological, molecular and functional changes and become reactive astrocytes. The changes associated with the reactive state can directly impact synaptic transmission and neuronal circuit activity, thereby potentially contributing to the pathological changes observed after ischemia.
On the other hand, resting microglia in the mature brain is a powerful weapon under
pathological activation. Following ischemia, as a result of rapid neuronal death, microglia migrates to damaged tissue to exert a neuroprotective effect by clearing dead tissue, inhibiting cytotoxic neuronal damage and releasing neuroprotective growth factors. However, microglial activation triggers iNOS production, which, together with the parallel appearance of reactive astrocytes, results in massive iNOS expression observed relatively late after an ischemic episode (after approximately 24 h) and generation of ROS/RNS.
Pathological glial activation escapes endogenous control and turns into an autoaggressive pathomechanism that contributes to secondary neuronal damage occurring after brain ischemia. Leukocytes accumulating at the site of neuronal necrosis act as an additional barrier to this cascade of destructive events by secreting cytokines, including interferon-γ and interleukins ( Figure 2 ).
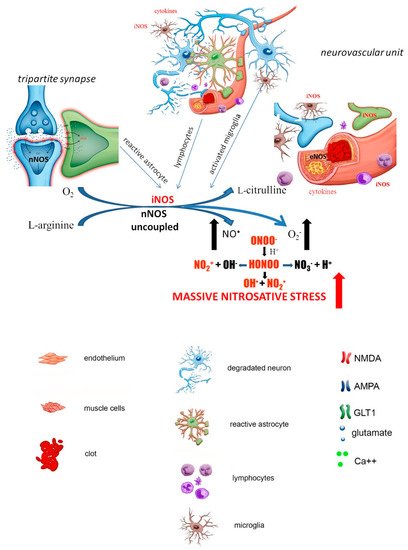
Reassuming, ischemic brain injury involves a complex interaction between leukocytes, glia, neurons and the endothelium, which form a vicious chain of events that results in massive and continuous production of iNOS resulting is subsequent generation of ROS/RNS. The pathological events associated with ischemic brain injury involve energy failure, oxidative stress, acidosis, disruption of ion homeostasis, neuronal cell excitotoxicity or inflammation and evolve progressively over time. The early and late stages of ischemia described above concern not only sequential NO • production but also immunological responses (which are in part related to NO • generation) described elsewhere in more details.
2. NO-Dependent Factors Aggravating Ischemic Cascade
In addition to guanylyl cyclase activation and subsequent initialization of cGMP production (or pathological generation of ROS/RNS), NO•-mediated processes control the functioning of many proteins and genes expression. Among them, s-nitrosylation and HIF-1α stabilization seem to be of importance in pathophysiology of brain ischemia.
The NO•-mediated S-nitrosylation process is a redox-based posttranslational modification that modulates protein function and activity. S-nitrosylation is the chemical reaction of an NO• moiety with the sulfhydryl groups of target proteins, which leads to the formation of S-nitrosothiols (R-SNO), producing S-nitrosylated proteins (SNO-proteins).
Aberrant S-nitrosylation occurs as a consequence of exacerbated nitrosative stress via the excessive production of NO•, which nitrosylates cysteine thiols with only partial SNO motifs or located more distant from the NO• source. These aberrantly S-nitrosylated proteins may contribute to pathological changes by triggering protein misfolding, mitochondrial dysfunction, transcriptional dysregulation, synaptic damage and neuronal injury. Two of the most important proteins that undergo aberrant S-nitrosylation in response to ischemic injury are GAPDH and matrix metalloproteinase 9 (MMP9). GAPDH has been implicated in neurotoxicity and neurodegeneration and regulates transcriptional activation, apoptosis initiation, ER to Golgi vesicle shuttling and fast axonal or axoplasmic transport. MMP9 belongs to the zinc metalloproteinase family, which is involved in the degradation of the extracellular matrix. The enzyme is acutely activated during ischemia and is selectively S-nitrosylated by NO• . HIF-1α is the primary mammalian transcription factor specifically regulated by hypoxia and plays an essential role in cellular and systemic O2 homeostasis by regulating the expression of genes important in tissue survival, that regulate glycolysis, erythropoiesis, angiogenesis or catecholamine metabolism.
The role of HIF-1α in ischemia remains inconclusive, but despite controversial
results the role of HIF-1α was shown to be of special importance in ischemia preconditioning and may either promote or prevent neuronal survival. Partially the pro-survival and pro-inflammatory roles in the ischemic brain might depend on the injury model, time point or cell type assessed.
3. Treatment Strategies
The advantage of NO•-based therapies is the possibility of modulating endogenous
mechanisms activated after cerebral ischemia with exogenously applied compounds. The objective is to promote neuroprotective outcomes and integrate cellular signaling pathways at different stages of brain damage. The intervention time, pharmacokinetics, pharmacodynamics and activities of the compounds are critical to successfully counteract the consequences of stroke.
The present knowledge indicates that the therapeutic window to reduce the pathological consequences of stroke, essentially neuronal damage, is estimated as 0–6 h for primary interventions and may extend up to 24 h poststroke.
As discussed above, NO-mediated actions clearly indicate that the activation of
eNOS contributes to proper vasodilation, exerts antioxidant, anti-inflammatory and antiatherogenic effects, and regulates glucose uptake and insulin sensitivity, thus exerting protective effects in stroke. In contrast, the inhibition of nNOS and iNOS, the main generators of free radicals, alternatively elimination of free radicals, may counteract neurodegeneration.


