 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Haruki Odaka | + 4130 word(s) | 4130 | 2021-06-11 05:15:43 | | | |
| 2 | Lily Guo | Meta information modification | 4130 | 2021-06-16 08:35:54 | | |
Video Upload Options
The function of the brain-derived neurotrophic factor (BDNF) via activation through its high-affinity receptor Tropomyosin receptor kinase B (TrkB) has a pivotal role in cell differentiation, cell survival, synaptic plasticity, and both embryonic and adult neurogenesis in central nervous system neurons. A number of studies have demonstrated the possible involvement of altered expression and action of the BDNF/TrkB signaling in the pathogenesis of neurodegenerative diseases, including Alzheimer’s disease (AD). In this review, we introduce an essential role of the BDNF and its downstream signaling in neural function. We also review the current evidence on the deregulated the BDNF signaling in the pathophysiology of AD at gene, mRNA, and protein levels. Further, we discuss a potential usefulness of small compounds, including flavonoids, which can stimulate BDNF-related signaling as a BDNF-targeting therapy.
1. Introduction
2. The BDNF and Its Intracellular Signaling
2.1. The BDNF/TrkB Signaling-Mediated Pathways
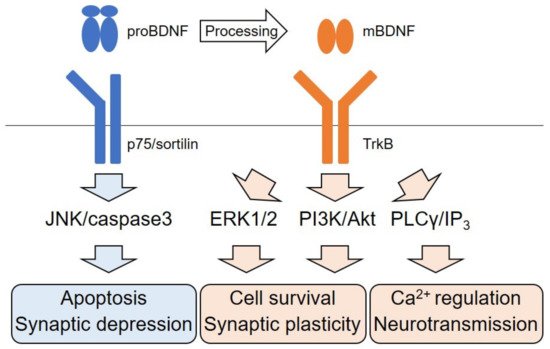
2.2. Dichotomic Effects of ERK Signaling Activation on Neuronal Survival
2.3. Neurotransmitter Release Through the TrkB/PLCγ Pathway
2.4. p75 Signaling Pathway
3. BDNF Signaling in AD
3.1. Downregulation of the BDNF in AD
3.2. Imbalance of proBDNF Transformation into Mature BDNF
3.3. Types of TrkB Receptors and AD
3.4. The BDNF Polymorphism and AD
3.5. Pathogenic Role of the BDNF in AD
3.6. The BDNF, Dysregulation of the Cholinergic System, and Dementia in Down Syndrome
References
- Numakawa, T.; Odaka, H. Brain-derived neurotrophic factor and neurogenesis. In Factors Affecting Neurodevelopment: Genetics, Neurology, Behavior, and Diet; Martin, C., Preedy, V., Rajendram, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2021.
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560.
- Garzon, D.; Yu, G.; Fahnestock, M. A new brain-derived neurotrophic factor transcript and decrease in brain-derived neurotrophic factor transcripts 1, 2 and 3 in Alzheimer’s disease parietal cortex. J. Neurochem. 2002, 82, 1058–1064.
- Phillips, H.S.; Hains, J.M.; Armanini, M.; Laramee, G.R.; Johnson, S.A.; Winslow, J.W. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron 1991, 7, 695–702.
- Hock, C.; Heese, K.; Hulette, C.; Rosenberg, C.; Otten, U. Region-specific neurotrophin imbalances in Alzheimer disease: Decreased levels of brain-derived neurotrophic factor and increased levels of nerve growth factor in hippocampus and cortical areas. Arch. Neurol. 2000, 57, 846–851.
- Garzon, D.J.; Fahnestock, M. Oligomeric amyloid decreases basal levels of brain-derived neurotrophic factor (BDNF) mRNA via specific downregulation of BDNF transcripts IV and V in differentiated human neuroblastoma cells. J. Neurosci. 2007, 27, 2628–2635.
- Rohe, M.; Synowitz, M.; Glass, R.; Paul, S.M.; Nykjaer, A.; Willnow, T.E. Brain-derived neurotrophic factor reduces amyloidogenic processing through control of SORLA gene expression. J. Neurosci. 2009, 29, 15472–15478.
- Elliott, E.; Atlas, R.; Lange, A.; Ginzburg, I. Brain-derived neurotrophic factor induces a rapid dephosphorylation of tau protein through a PI-3 Kinase signalling mechanism. Eur. J. Neurosci. 2005, 22, 1081–1089.
- Fleitas, C.; Piñol-Ripoll, G.; Marfull, P.; Rocandio, D.; Ferrer, I.; Rampon, C.; Egea, J.; Espinet, C. proBDNF is modified by advanced glycation end products in Alzheimer’s disease and causes neuronal apoptosis by inducing p75 neurotrophin receptor processing. Mol. Brain 2018, 11, 68.
- Wurzelmann, M.; Romeika, J.; Sun, D. Therapeutic potential of brain-derived neurotrophic factor (BDNF) and a small molecular mimics of BDNF for traumatic brain injury. Neural Regen. Res. 2017, 12, 7–12.
- Numakawa, T.; Suzuki, S.; Kumamaru, E.; Adachi, N.; Richards, M.; Kunugi, H. BDNF function and intracellular signaling in neurons. Histol. Histopathol. 2010, 25, 237–258.
- Lu, B.; Nagappan, G.; Guan, X.; Nathan, P.J.; Wren, P. BDNF-based synaptic repair as a disease-modifying strategy for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 401–416.
- Xue, Y.; Liang, H.; Yang, R.; Deng, K.; Tang, M.; Zhang, M. The role of pro- and mature neurotrophins in the depression. Behav. Brain Res. 2021, 404, 113162.
- Numakawa, T.; Odaka, H.; Adachi, N. Actions of Brain-Derived Neurotrophin Factor in the Neurogenesis and Neuronal Function, and Its Involvement in the Pathophysiology of Brain Diseases. Int. J. Mol. Sci. 2018, 19, 3650.
- Guo, W.; Nagappan, G.; Lu, B. Differential effects of transient and sustained activation of BDNF-TrkB signaling. Dev. Neurobiol. 2018, 78, 647–659.
- Sharma, P.; Kumar, A.; Singh, D. Dietary Flavonoids Interaction with CREB-BDNF Pathway: An Unconventional Approach for Comprehensive Management of Epilepsy. Curr. Neuropharmacol. 2019, 17, 1158–1175.
- Ohira, K.; Hayashi, M. A new aspect of the TrkB signaling pathway in neural plasticity. Curr. Neuropharmacol. 2009, 7, 276–285.
- Numakawa, T.; Kumamaru, E.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Kunugi, H. Glucocorticoid receptor interaction with TrkB promotes BDNF-triggered PLC-gamma signaling for glutamate release via a glutamate transporter. Proc. Natl. Acad. Sci. USA 2009, 106, 647–652.
- Kumamaru, E.; Numakawa, T.; Adachi, N.; Yagasaki, Y.; Izumi, A.; Niyaz, M.; Kudo, M.; Kunugi, H. Glucocorticoid prevents brain-derived neurotrophic factor-mediated maturation of synaptic function in developing hippocampal neurons through reduction in the activity of mitogen-activated protein kinase. Mol. Endocrinol. 2008, 22, 546–558.
- Yang, J.R.; Ren, T.T.; Lan, R.; Qin, X.Y. Tea polyphenols attenuate staurosporine-induced cytotoxicity and apoptosis by modulating BDNF-TrkB/Akt and Erk1/2 signaling axis in hippocampal neurons. IBRO Rep. 2020, 8, 115–121.
- Li, Y.; Xiang, L.; Wang, C.; Song, Y.; Miao, J.; Miao, M. Protection against acute cerebral ischemia/reperfusion injury by Leonuri Herba Total Alkali via modulation of BDNF-TrKB-PI3K/Akt signaling pathway in rats. Biomed. Pharmacother. 2021, 133, 111021.
- Ma, Z.; Liu, K.; Li, X.R.; Wang, C.; Liu, C.; Yan, D.Y.; Deng, Y.; Liu, W.; Xu, B. Alpha-synuclein is involved in manganese-induced spatial memory and synaptic plasticity impairments via TrkB/Akt/Fyn-mediated phosphorylation of NMDA receptors. Cell Death Dis. 2020, 11, 834.
- Numakawa, Y.; Matsumoto, T.; Yokomaku, D.; Taguchi, T.; Niki, E.; Hatanaka, H.; Kunugi, H.; Numakawa, T. 17beta-estradiol protects cortical neurons against oxidative stress-induced cell death through reduction in the activity of mitogen-activated protein kinase and in the accumulation of intracellular calcium. Endocrinology 2007, 148, 627–637.
- Huang, C.F.; Liu, S.H.; Su, C.C.; Fang, K.M.; Yen, C.C.; Yang, C.Y.; Tang, F.C.; Liu, J.M.; Wu, C.C.; Lee, K.I.; et al. Roles of ERK/Akt signals in mitochondria-dependent and endoplasmic reticulum stress-triggered neuronal cell apoptosis induced by 4-methyl-2,4-bis(4-hydroxyphenyl)pent-1-ene, a major active metabolite of bisphenol A. Toxicology 2021, 455, 152764.
- Zhang, Z.; Fan, J.; Ren, Y.; Zhou, W.; Yin, G. The release of glutamate from cortical neurons regulated by BDNF via the TrkB/Src/PLC-γ1 pathway. J. Cell Biochem. 2013, 114, 144–151.
- Suelves, N.; Miguez, A.; López-Benito, S.; Barriga, G.G.; Giralt, A.; Alvarez-Periel, E.; Arévalo, J.C.; Alberch, J.; Ginés, S.; Brito, V. Early Downregulation of p75(NTR) by Genetic and Pharmacological Approaches Delays the Onset of Motor Deficits and Striatal Dysfunction in Huntington’s Disease Mice. Mol. Neurobiol. 2019, 56, 935–953.
- Asuni, G.P.; Speidell, A.; Mocchetti, I. Neuronal apoptosis induced by morphine withdrawal is mediated by the p75 neurotrophin receptor. J. Neurochem. 2021.
- Wong, L.W.; Chong, Y.S.; Lin, W.; Kisiswa, L.; Sim, E.; Ibáñez, C.F.; Sajikumar, S. Age-related changes in hippocampal-dependent synaptic plasticity and memory mediated by p75 neurotrophin receptor. Aging Cell 2021, 20, e13305.
- Wong, L.W.; Tann, J.Y.; Ibanez, C.F.; Sajikumar, S. The p75 Neurotrophin Receptor Is an Essential Mediator of Impairments in Hippocampal-Dependent Associative Plasticity and Memory Induced by Sleep Deprivation. J. Neurosci. 2019, 39, 5452–5465.
- Du, Y.; Wu, H.T.; Qin, X.Y.; Cao, C.; Liu, Y.; Cao, Z.Z.; Cheng, Y. Postmortem Brain, Cerebrospinal Fluid, and Blood Neurotrophic Factor Levels in Alzheimer’s Disease: A Systematic Review and Meta-Analysis. J. Mol. Neurosci. 2018, 65, 289–300.
- Keifer, J. Comparative Genomics of the BDNF Gene, Non-Canonical Modes of Transcriptional Regulation, and Neurological Disease. Mol. Neurobiol. 2021, 58, 2851–2861.
- Rosa, E.; Mahendram, S.; Ke, Y.D.; Ittner, L.M.; Ginsberg, S.D.; Fahnestock, M. Tau downregulates BDNF expression in animal and cellular models of Alzheimer’s disease. Neurobiol. Aging 2016, 48, 135–142.
- Wang, M.; Xie, Y.; Qin, D. Proteolytic cleavage of proBDNF to mBDNF in neuropsychiatric and neurodegenerative diseases. Brain Res. Bull. 2021, 166, 172–184.
- Niculescu, D.; Michaelsen-Preusse, K.; Güner, Ü.; van Dorland, R.; Wierenga, C.J.; Lohmann, C. A BDNF-Mediated Push-Pull Plasticity Mechanism for Synaptic Clustering. Cell Rep. 2018, 24, 2063–2074.
- Ledesma, M.D.; Da Silva, J.S.; Crassaerts, K.; Delacourte, A.; De Strooper, B.; Dotti, C.G. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer’s disease brains. EMBO Rep. 2000, 1, 530–535.
- Cai, M.; Jung, I.; Kwon, H.; Cho, E.; Jeon, J.; Yun, J.; Lee, Y.C.; Kim, D.H.; Ryu, J.H. Spinosin Attenuates Alzheimer’s Disease-Associated Synaptic Dysfunction via Regulation of Plasmin Activity. Biomol. Ther. 2020, 28, 131–136.
- Fabbro, S.; Seeds, N.W. Plasminogen activator activity is inhibited while neuroserpin is up-regulated in the Alzheimer disease brain. J. Neurochem. 2009, 109, 303–315.
- Gerenu, G.; Martisova, E.; Ferrero, H.; Carracedo, M.; Rantamäki, T.; Ramirez, M.J.; Gil-Bea, F.J. Modulation of BDNF cleavage by plasminogen-activator inhibitor-1 contributes to Alzheimer’s neuropathology and cognitive deficits. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 991–1001.
- Kaminari, A.; Giannakas, N.; Tzinia, A.; Tsilibary, E.C. Overexpression of matrix metalloproteinase-9 (MMP-9) rescues insulin-mediated impairment in the 5XFAD model of Alzheimer’s disease. Sci. Rep. 2017, 7, 683.
- Hu, T.; Zhou, Y.; Lu, J.; Xia, P.; Chen, Y.; Cao, X.; Pei, G. A novel rhamnoside derivative PL402 up-regulates matrix metalloproteinase 3/9 to promote Aβ degradation and alleviates Alzheimer’s-like pathology. Aging 2020, 12, 481–501.
- Song, J.; Wu, C.; Korpos, E.; Zhang, X.; Agrawal, S.M.; Wang, Y.; Faber, C.; Schäfers, M.; Körner, H.; Opdenakker, G.; et al. Focal MMP-2 and MMP-9 activity at the blood-brain barrier promotes chemokine-induced leukocyte migration. Cell Rep. 2015, 10, 1040–1054.
- Agrawal, S.; Anderson, P.; Durbeej, M.; van Rooijen, N.; Ivars, F.; Opdenakker, G.; Sorokin, L.M. Dystroglycan is selectively cleaved at the parenchymal basement membrane at sites of leukocyte extravasation in experimental autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1007–1019.
- Hannocks, M.J.; Zhang, X.; Gerwien, H.; Chashchina, A.; Burmeister, M.; Korpos, E.; Song, J.; Sorokin, L. The gelatinases, MMP-2 and MMP-9, as fine tuners of neuroinflammatory processes. Matrix Biol. 2019, 75–76, 102–113.
- Jerónimo-Santos, A.; Vaz, S.H.; Parreira, S.; Rapaz-Lérias, S.; Caetano, A.P.; Buée-Scherrer, V.; Castrén, E.; Valente, C.A.; Blum, D.; Sebastião, A.M.; et al. Dysregulation of TrkB Receptors and BDNF Function by Amyloid-β Peptide is Mediated by Calpain. Cereb. Cortex 2015, 25, 3107–3121.
- Kemppainen, S.; Rantamäki, T.; Jerónimo-Santos, A.; Lavasseur, G.; Autio, H.; Karpova, N.; Kärkkäinen, E.; Stavén, S.; Vicente Miranda, H.; Outeiro, T.F.; et al. Impaired TrkB receptor signaling contributes to memory impairment in APP/PS1 mice. Neurobiol. Aging 2012, 33, 1122.e23–1122.e39.
- Ferrer, I.; Marín, C.; Rey, M.J.; Ribalta, T.; Goutan, E.; Blanco, R.; Tolosa, E.; Martí, E. BDNF and full-length and truncated TrkB expression in Alzheimer disease. Implications in therapeutic strategies. J. Neuropathol. Exp. Neurol. 1999, 58, 729–739.
- Urbina-Varela, R.; Soto-Espinoza, M.I.; Vargas, R.; Quiñones, L.; Del Campo, A. Influence of BDNF Genetic Polymorphisms in the Pathophysiology of Aging-related Diseases. Aging Dis. 2020, 11, 1513–1526.
- Chen, Z.Y.; Patel, P.D.; Sant, G.; Meng, C.X.; Teng, K.K.; Hempstead, B.L.; Lee, F.S. Variant brain-derived neurotrophic factor (BDNF) (Met66) alters the intracellular trafficking and activity-dependent secretion of wild-type BDNF in neurosecretory cells and cortical neurons. J. Neurosci. 2004, 24, 4401–4411.
- Hao, R.; Qi, Y.; Hou, D.N.; Ji, Y.Y.; Zheng, C.Y.; Li, C.Y.; Yung, W.H.; Lu, B.; Huang, Y. BDNF val66met Polymorphism Impairs Hippocampal Long-Term Depression by Down-Regulation of 5-HT3 Receptors. Front. Cell. Neurosci. 2017, 11, 306.
- Egan, M.F.; Kojima, M.; Callicott, J.H.; Goldberg, T.E.; Kolachana, B.S.; Bertolino, A.; Zaitsev, E.; Gold, B.; Goldman, D.; Dean, M.; et al. The BDNF val66met polymorphism affects activity-dependent secretion of BDNF and human memory and hippocampal function. Cell 2003, 112, 257–269.
- Lim, Y.Y.; Villemagne, V.L.; Laws, S.M.; Ames, D.; Pietrzak, R.H.; Ellis, K.A.; Harrington, K.; Bourgeat, P.; Bush, A.I.; Martins, R.N.; et al. Effect of BDNF Val66Met on memory decline and hippocampal atrophy in prodromal Alzheimer’s disease: A preliminary study. PLoS ONE 2014, 9, e86498.
- Boots, E.A.; Schultz, S.A.; Clark, L.R.; Racine, A.M.; Darst, B.F.; Koscik, R.L.; Carlsson, C.M.; Gallagher, C.L.; Hogan, K.J.; Bendlin, B.B.; et al. BDNF Val66Met predicts cognitive decline in the Wisconsin Registry for Alzheimer’s Prevention. Neurology 2017, 88, 2098–2106.
- Fukumoto, N.; Fujii, T.; Combarros, O.; Kamboh, M.I.; Tsai, S.J.; Matsushita, S.; Nacmias, B.; Comings, D.E.; Arboleda, H.; Ingelsson, M.; et al. Sexually dimorphic effect of the Val66Met polymorphism of BDNF on susceptibility to Alzheimer’s disease: New data and meta-analysis. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2010, 153, 235–242.
- He, X.M.; Zhang, Z.X.; Zhang, J.W.; Zhou, Y.T.; Tang, M.N.; Wu, C.B.; Hong, Z. Lack of association between the BDNF gene Val66Met polymorphism and Alzheimer disease in a Chinese Han population. Neuropsychobiology 2007, 55, 151–155.
- Matrone, C.; Ciotti, M.T.; Mercanti, D.; Marolda, R.; Calissano, P. NGF and BDNF signaling control amyloidogenic route and Abeta production in hippocampal neurons. Proc. Natl. Acad. Sci. USA 2008, 105, 13139–13144.
- Wang, Z.H.; Xiang, J.; Liu, X.; Yu, S.P.; Manfredsson, F.P.; Sandoval, I.M.; Wu, S.; Wang, J.Z.; Ye, K. Deficiency in BDNF/TrkB Neurotrophic Activity Stimulates δ-Secretase by Upregulating C/EBPβ in Alzheimer’s Disease. Cell Rep. 2019, 28, 655–669.e5.
- Braun, D.J.; Kalinin, S.; Feinstein, D.L. Conditional Depletion of Hippocampal Brain-Derived Neurotrophic Factor Exacerbates Neuropathology in a Mouse Model of Alzheimer’s Disease. ASN Neuro 2017, 9, 1759091417696161.
- Aytan, N.; Choi, J.K.; Carreras, I.; Crabtree, L.; Nguyen, B.; Lehar, M.; Blusztajn, J.K.; Jenkins, B.G.; Dedeoglu, A. Protective effects of 7,8-dihydroxyflavone on neuropathological and neurochemical changes in a mouse model of Alzheimer’s disease. Eur. J. Pharmacol. 2018, 828, 9–17.
- Castello, N.A.; Green, K.N.; LaFerla, F.M. Genetic knockdown of brain-derived neurotrophic factor in 3xTg-AD mice does not alter Aβ or tau pathology. PLoS ONE 2012, 7, e39566.
- Devi, L.; Ohno, M. TrkB reduction exacerbates Alzheimer’s disease-like signaling aberrations and memory deficits without affecting β-amyloidosis in 5XFAD mice. Transl. Psychiatry 2015, 5, e562.
- De Pins, B.; Cifuentes-Díaz, C.; Farah, A.T.; López-Molina, L.; Montalban, E.; Sancho-Balsells, A.; López, A.; Ginés, S.; Delgado-García, J.M.; Alberch, J.; et al. Conditional BDNF Delivery from Astrocytes Rescues Memory Deficits, Spine Density, and Synaptic Properties in the 5xFAD Mouse Model of Alzheimer Disease. J. Neurosci. 2019, 39, 2441–2458.
- Duan, X.; Li, Y.; Xu, F.; Ding, H. Study on the neuroprotective effects of Genistein on Alzheimer’s disease. Brain Behav. 2021, 11, e02100.
- Cheng, Y.J.; Lin, C.H.; Lane, H.Y. Involvement of Cholinergic, Adrenergic, and Glutamatergic Network Modulation with Cognitive Dysfunction in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2283.
- Shekari, A.; Fahnestock, M. Retrograde axonal transport of BDNF and proNGF diminishes with age in basal forebrain cholinergic neurons. Neurobiol. Aging 2019, 84, 131–140.
- Lv, J.; Lu, C.; Jiang, N.; Wang, H.; Huang, H.; Chen, Y.; Li, Y.; Liu, X. Protective effect of ginsenoside Rh2 on scopolamine-induced memory deficits through regulation of cholinergic transmission, oxidative stress and the ERK-CREB-BDNF signaling pathway. Phytother. Res. 2021, 35, 337–345.
- Sun, Y.; Zhao, Z.; Li, Q.; Wang, C.; Ge, X.; Wang, X.; Wang, G.; Qin, Y. Dl-3-n-butylphthalide regulates cholinergic dysfunction in chronic cerebral hypoperfusion rats. J. Int. Med. Res. 2020, 48, 300060520936177.
- Shin, J.; Kong, C.; Lee, J.; Choi, B.Y.; Sim, J.; Koh, C.S.; Park, M.; Na, Y.C.; Suh, S.W.; Chang, W.S.; et al. Focused ultrasound-induced blood-brain barrier opening improves adult hippocampal neurogenesis and cognitive function in a cholinergic degeneration dementia rat model. Alzheimer’s Res. Ther. 2019, 11, 110.
- Isacson, O.; Seo, H.; Lin, L.; Albeck, D.; Granholm, A.C. Alzheimer’s disease and Down’s syndrome: Roles of APP, trophic factors and ACh. Trends Neurosci. 2002, 25, 79–84.
- Seo, H.; Isacson, O. Abnormal APP, cholinergic and cognitive function in Ts65Dn Down’s model mice. Exp. Neurol. 2005, 193, 469–480.
- Velazquez, R.; Ash, J.A.; Powers, B.E.; Kelley, C.M.; Strawderman, M.; Luscher, Z.I.; Ginsberg, S.D.; Mufson, E.J.; Strupp, B.J. Maternal choline supplementation improves spatial learning and adult hippocampal neurogenesis in the Ts65Dn mouse model of Down syndrome. Neurobiol. Dis. 2013, 58, 92–101.
- Giacomini, A.; Stagni, F.; Emili, M.; Uguagliati, B.; Rimondini, R.; Bartesaghi, R.; Guidi, S. Timing of Treatment with the Flavonoid 7,8-DHF Critically Impacts on Its Effects on Learning and Memory in the Ts65Dn Mouse. Antioxidants 2019, 8, 163.
- Stagni, F.; Giacomini, A.; Guidi, S.; Emili, M.; Uguagliati, B.; Salvalai, M.E.; Bortolotto, V.; Grilli, M.; Rimondini, R.; Bartesaghi, R. A flavonoid agonist of the TrkB receptor for BDNF improves hippocampal neurogenesis and hippocampus-dependent memory in the Ts65Dn mouse model of DS. Exp. Neurol. 2017, 298, 79–96.


