 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Takashi Obama | + 2059 word(s) | 2059 | 2020-11-25 07:24:17 | | | |
| 2 | Peter Tang | Meta information modification | 2059 | 2020-12-08 14:06:28 | | |
Video Upload Options
Neutrophil extracellular traps (NETs) significantly contribute to various pathophysiological conditions, including cardiovascular diseases. NET formation in the vasculature exhibits inflammatory and thrombogenic activities on the endothelium. NETs are induced by various stimulants such as exogenous damage-associated molecular patterns (DAMPs).
1. Introduction
Neutrophil extracellular traps (NETs) were initially considered to be one of the first line responses of the immune system against infected bacteria [1], extensive studies have revealed that NET formation is associated with the initiation and progression of various noninfectious diseases [2][3]. In cardiovascular diseases (CVDs), NETs have been found in vascular lesions such as atherosclerotic plaques and thrombi [4].
Neutrophils or polymorphonuclear leukocytes (PMNs) have a short lifespan and are highly susceptible to activation. The human promyelocytic leukemia cell line, HL-60, is widely used in the in vitro analysis of NETs. HL-60 cells can be differentiated into neutrophil-like cells using various pharmacological stimulants, such as all-trans retinoic acid, dimethyl sulfoxide, and dimethylformamide. HL-60-derived neutrophil-like cells reveal NET formation characterized by CD11b expression, DNA release, oxidative burst, and histone citrullination; however, these responses are more pronounced in PMNs than in HL-60 [5]. Phorbol myristate acetate (PMA), a potent protein kinase C activator, is a well-defined NET-inducer [6]. In addition to PMA, several cytokines and chemokines have been reported to induce NET formation, including TNFα, IL-8 [1], IL-18 [7], CXCL7 [8], complement (C5a) [9], and interferons [10]. Moreover, NET formation is primed by crystals of monosodium urate [11][12] and cholesterol [13]. High concentrations of glucose induce NETosis [14], indicating that NET formation is increased in patients with type 2 diabetes [15] and diabetic retinopathy [16]. Furthermore, nicotine also induces NET formation [17], and PMNs from smokers are more susceptible to PMA-induced NET formation than those from nonsmokers [18].
NET release can be classified into two types: (1) "suicidal NETs", which proceed in 3–4 h, and induce DNA release concomitantly by rupturing neutrophils to cause cell death (NETosis); and (2) "vital NETs", which release DNA via vesicles into the extracellular space within 60 min, without causing cell death [2][19]. Notably, the term "NETosis" is applicable only when experimental evidence clearly supports cell death upon NETting [20]. While NET formation is elicited via various signaling pathways involving ROS production [21], the two types of NETs are mediated by different sources of ROS. The Raf-MEK-ERK pathway stimulates NADPH oxidase-mediated ROS production in the process of suicidal NETs [22], whereas mitochondrial ROS is mainly required in vital NETs [17][23]. Chromatin decondensation associated with fragmentation of the nucleus is an important step that releases DNA into the extracellular spaces. MPO and proteases, particularly NE, translocate from azurophilic granules to the nucleus to promote disintegration of the chromatin [24], leading to histone degradation [25]. Gasdermin D is a pore-forming protein that was initially identified as a crucial factor for pyroptotic cell death of macrophages [26][27]. NE-dependent cleavage of gasdermin D activates itself, which in turn plays a pivotal role in suicidal NETs by expanding granules and nucleus, and eventually ruptures the plasma membrane [28][29].
DNA strands released from neutrophils are decorated by proteins derived from the cytoplasm, azurophil granules, and nuclei. Intracellular ROS production activates peptidylarginine deiminase 4 (PAD4), which catalyzes the posttranslational modification of positively charged arginine residues of histones to neutral citrulline residues. Previous studies have demonstrated the necessity of PAD4 activity for NET formation, whereas most recent studies that have compared the process of NET formation in mouse and human neutrophils, in addition to HL-60-derived neutrophils, have revealed that PAD4 enzymatic activity is crucial for efficient DNA decondensation [30][31].
2. NET-Related Receptors
Membrane receptor-mediated signaling pathways lead to NET formation via ROS production and PAD4 activation [25]. In general, toll-like receptors (TLRs) on the plasma membrane of neutrophils play distinctive roles in NET formation in the presence of bacterial pathogen-derived stimuli, including lipopolysaccharides [25].
The receptor for advanced glycation end products (RAGE) is expressed on human neutrophils [32]. High-mobility group box 1 (HMGB1), a DNA-binding protein secreted from macrophages and monocytes, which acts as a damage-associated molecular pattern (DAMP) to mediate thrombosis [33], is expressed on activated platelets. It has been reported that binding of HMGB1 to RAGE on neutrophils induces NET formation [32].
Fcγ receptors expressed on PMNs participate in the induction of ROS production as well as recognition of antibody-opsonized pathogens [34]. The immune complex increases NET formation in murine neutrophils expressing human FcγRIIa (CD32a), but not FcγRIIIb (CD16b) [35]; however, among the antibody receptors expressed by human neutrophils, only FcγRIIIb (CD16b) is responsible for NET formation in response to cross-linking antibodies [36]. Experimental differences, including cell types and stimulants, may be responsible for these differences in findings. Interestingly, TLR7/8 activation leads to proteolytic cleavage of FcγRIIa, thereby shifting neutrophils from phagocytosis of immune complexes to NET formation [37]. Clinically relevant evidence has revealed that neutrophils from patients with systemic lupus erythematosus (SLE) demonstrate similar cleavage of FcγRIIa related to that of neutrophil activation [37].
3. NETs and Cardiovascular Diseases
Several studies regarding the pathophysiological roles of NETs have elucidated the implication of NETs [38] with coronary artery [39][40] and venous thrombosis [41]. Immunothrombosis and NET-induced thrombosis are important and widely studied phenomena [42][43][44]. Plaque rupture and plaque erosion are two major causes of atherothrombosis. Plaques prone to rupture are characterized by a large lipid core, thin fibrous cap, and numerous macrophages, but few smooth muscle cells (SMCs), which form red fibrin-rich thrombi. Plaque erosion, which causes white platelet-rich thrombi, is associated with enriched collagen, abundant SMCs, and little accumulation of lipids and foam cells [45] (Figure 1). Neutrophils infiltrate culprit lesions, including both ruptured and eroded plaques [46], via NET formation [47]. By activating neutrophils, circulating MPO levels are increased in patients with acute coronary syndrome [48]. Lipid-lowering pharmacotherapy with statins reduces plasma cholesterol levels and suppresses lipid accumulation in the lesions, thus exerting stabilizing effects on rupture-prone atherosclerotic plaques with reduced inflammation in the vascular tissue [45][49][50][51]; however, a remarkable number of patients have experienced cardiovascular events even after achieving a marked reduction in their cholesterol levels by statin therapy; accordingly, the underlying mechanism for superficial erosion has gained immense attention [52]. Presently, NETs are regarded as pivotal contributors in the formation of thrombi during superficial erosion [53][54]. This is in accordance with the observations that MPO levels in plasma and the density of MPO-positive cells in thrombi are elevated in acute coronary disease patients with eroded culprit plaques, but not in patients with ruptured culprit plaques [55][56][57].
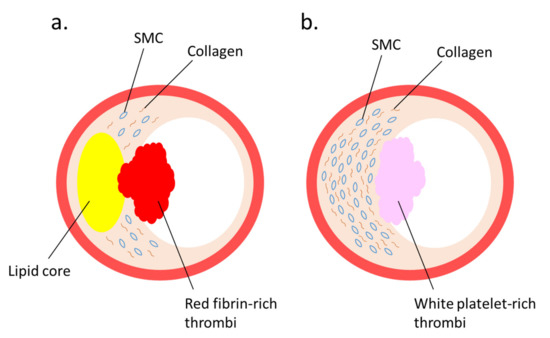
Figure 1. Two major lesions that lead to thrombus formation: (a) plaque rupture and (b) plaque erosion—are illustrated. (a) A vulnerable plaque is characterized by a large lipid core covered by a thin fibrous cap. When the fibrous cap is ruptured, red thrombus is produced. (b) A plaque erosion is characterized by a thickened intima enriched with smooth muscle cells (SMCs) and collagen fibers. When endothelial cells are injured, white-platelet-rich thrombus is produced.
NETs have been implicated in metabolic abnormalities such as diabetes. Neutrophils isolated from type 1 and type 2 diabetes are sensitive to NET formation, and the accelerated NETs impair wound healing [19]. Hyperglycemia is correlated with elevated white blood cells and reduced high-density lipoprotein (HDL) cholesterol levels. In diabetic mice, raising the levels of functional HDL, which accepts cholesterol from peripheral tissues, leads to regression of atherosclerosis, associated with decreased plaque inflammation and reduced NET formation in the plaque [58].
The mechanistic link between NET formation and atherosclerosis/thrombosis has been widely investigated. NETs evoke inflammatory responses of immune cells to upregulate inflammatory cytokines [59]. NETs and their components not only damage ECs directly [60][61], but also trigger activation of ECs to form a scaffold, leading to thrombosis. In pathophysiological conditions, including vascular diseases, ECs lose their endothelial phenotype and then acquire a mesenchymal phenotype, which is characterized by the expression of fibroblastic markers [62]. Endothelial-to-mesenchymal transition (EndMT) is involved in the progression of atherosclerosis [62][63][64]. In general, NETs are internalized into ECs through RAGE via clathrin-dependent endocytosis; however, when excess amounts of NETs remain on the extracellular surface, it induces EndMT of ECs to loosen cell–cell contact, in which NE acts as a proteolytic enzyme of VE-cadherin [65].
4. Modified LDL and NET Formation
Oxidatively modified low-density lipoprotein (oxLDL) has been physiologically defined as a subpopulation of LDL that comprises various oxidative modifications in the protein components and oxidized lipids, which could act as DAMPs. oxLDL has been recognized as a crucial initiator and accelerator of atherosclerosis through foam cell formation by macrophages; however, recent studies have demonstrated that oxLDL stimulates neutrophils to induce NET formation and enhance NET-mediated inflammatory responses in vascular endothelial cells.
OxLDL acts as a DAMP and triggers sterile inflammatory responses to promote CVDs [6]. Infiltration of neutrophils into arteries during the early stages of atherosclerosis has been observed in hypercholesterolemic mice [66]. Numerous studies, on mice and humans, have provided evidence for the contribution of neutrophils to early atherosclerosis [67][68]. We have demonstrated the appearance of oxLDL in vascular tissues and in circulation even prior to atherosclerotic lesion development in apoE-KO mice [69]. These findings prompted us to examine whether oxLDL influences NET formation and subsequent endothelial inflammatory responses. PMA-induced NET formation in both HL-60-derived neutrophils and human PMNs is remarkably accelerated upon coincubation with oxLDL, but not with native LDL [70]. Moreover, extracellular components, after induction of NET formation by PMA with oxLDL, are capable of inducing morphological changes and MMP-1 expression in human aortic ECs (Figure 2). Presumably, neutrophils may play a pivotal role in the early stages of atherogenesis through the cooperative actions of oxLDL.
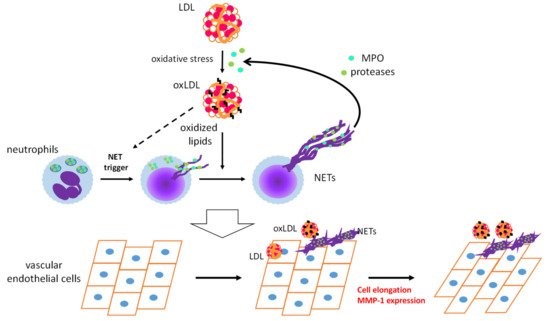
Figure 2. Schematic diagram of a feed-forward cascade that drives the neutrophil extracellular trap (NET)-induced endothelial inflammatory response. OxLDL strongly accelerates NET formation of activated neutrophils, while the same NET formation-inducing effect of oxLDL on resting neutrophils has not been observed. MPO and proteases released from neutrophils by NET formation could act on native low-density lipoprotein (LDL) to facilitate modification of LDL. Coexistence of NETs and oxLDL or native LDL promotes endothelial inflammation to cause cell elongation and enhanced MMP-1 production in endothelial cells [70]. MPO: myeloperoxidase, MMP-1: matrix metalloproteinase-1.
Intriguingly, NET-induced activation of human aortic ECs is enhanced upon coincubation with native LDL, suggesting that MPO and proteases released from neutrophils upon NET formation could act on native LDL and induce oxidative modification and/or degradation of LDL to produce modified proinflammatory LDL [11]. These phenomena, induced by the coexistence of NETs and native LDL or oxLDL, correspond to enhanced EndMT caused by the excess phagocytic capacity of ECs [65]. One possibility is that the cooperative action of oxLDL and NETs on ECs induces EndMT and subsequent neovascularization, which leads to atherosclerotic lesion formation [71].
Recently, a unique regulatory mechanism of lipoprotein-mediated vWF degradation has been described. LDL, but not HDL, has the ability to bind vWF and accelerate proteolytic cleavage of vWF by A disintegrin-like and metalloproteinase with thrombospondin type 1 motif 13 (ADAMTS13) in a concentration-dependent manner under shear stress; however, oxLDL competitively inhibits vWF proteolysis associated with native LDL. Furthermore, native LDL, but not oxLDL, inhibits adhesion of platelets to vWF on ECs under flow [72]. NET formation also inhibits the proteolytic activity of ADAMTS13 via PAD4-mediated citrullination [73]. ADAMTS13 is a specific protease for von Willebrand factor (vWF), known to cause thrombotic thrombocytopenic purpura [74]. vWF, a glycoprotein produced by endothelial cells (ECs), forms multimers and binds to collagen and Factor VIII, thus supporting platelet binding to the wound sites. vWF has been elucidated as one of the key factors in the development of NET-mediated endothelial damage and formation of thrombosis. Venous thrombi are characterized by red clots enriched with erythrocytes, because erythrocyte binding to vWF is promoted upon reduction of shear stress [75]. PAD4 released from NET-forming neutrophils citrullinates ADAMTS13; the modified ADAMTS13 loses its proteolytic activity against vWF, leading to the formation of thrombi after vessel injury via accumulation of platelets [73]. Considering that vWF functions as a scaffold for NETs, thrombosis, and platelets, these data indicate that oxLDL may enhance NET-mediated thrombosis formation via direct interaction with vWF (Figure 3).
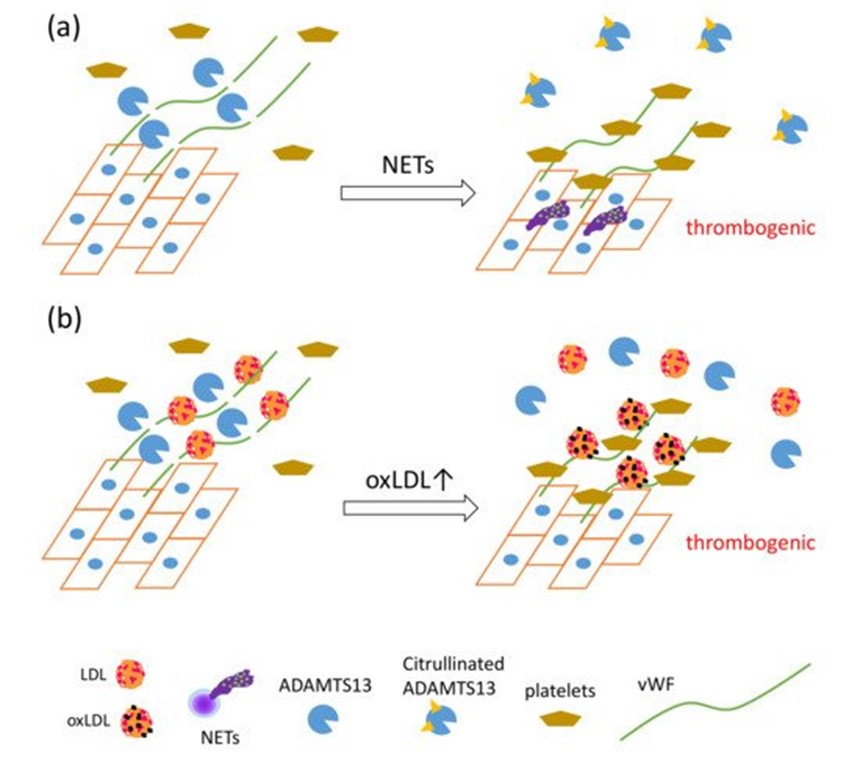
Figure 3. The synergistic actions of NET formation and oxLDL production presumably cause a proatherothrombotic status on the vascular endothelium. (a) PAD4 secreted by NET formation mediates citrullination of ADAMTS13, which inhibits the proteolytic activity of ADAMTS13 against von Willebrand factor (vWF), leading to increased vWF-platelet string formation on the endothelium [73]. (b) LDL, identified as a novel vWF-binding partner, accelerates the proteolytic cleavage of ultra-large vWF by ADAMTS13 under shear stress, and inhibits adhesion of platelets to ultra-large vWF. OxLDL, having lower ability to enhance proteolytic activity of ADAMTS13 than native LDL, competes with native LDL, which in turn increases platelet adhesion to ultra-large vWF, which could cause thrombus formation rich in vWF, platelets, and DNA [72].
References
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535.
- Castanheira, F.V.S.; Kubes, P. Neutrophils and NETs in modulating acute and chronic inflammation. Blood 2019, 133, 2178–2185.
- Sollberger, G.; Tilley, D.O.; Zychlinsky, A. Neutrophil extracellular traps: The biology of chromatin externalization. Dev. Cell 2018, 44, 542–553.
- Doring, Y.; Libby, P.; Soehnlein, O. Neutrophil extracellular traps participate in cardiovascular diseases: Recent experimental and clinical insights. Circ. Res. 2020, 126, 1228–1241.
- Manda-Handzlik, A.; Bystrzycka, W.; Wachowska, M.; Sieczkowska, S.; Stelmaszczyk-Emmel, A.; Demkow, U.; Ciepiela, O. The influence of agents differentiating HL-60 cells toward granulocyte-like cells on their ability to release neutrophil extracellular traps. Immunol. Cell. Biol. 2018, 96, 413–425.
- Hoppenbrouwers, T.; Autar, A.S.A.; Sultan, A.R.; Abraham, T.E.; van Cappellen, W.A.; Houtsmuller, A.B.; van Wamel, W.J.B.; van Beusekom, H.M.M.; van Neck, J.W.; de Maat, M.P.M. In vitro induction of NETosis: Comprehensive live imaging comparison and systematic review. PLoS ONE 2017, 12, e0176472.
- Kahlenberg, J.M.; Carmona-Rivera, C.; Smith, C.K.; Kaplan, M.J. Neutrophil extracellular trap-associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J. Immunol. 2013, 190, 1217–1226.
- Silvestre-Roig, C.; Braster, Q.; Wichapong, K.; Lee, E.Y.; Teulon, J.M.; Berrebeh, N.; Winter, J.; Adrover, J.M.; Santos, G.S.; Froese, A.; et al. Externalized histone H4 orchestrates chronic inflammation by inducing lytic cell death. Nature 2019, 569, 236–240.
- Yousefi, S.; Mihalache, C.; Kozlowski, E.; Schmid, I.; Simon, H.U. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell Death Differ. 2009, 16, 1438–1444.
- Bertin, F.R.; Rys, R.N.; Mathieu, C.; Laurance, S.; Lemarie, C.A.; Blostein, M.D. Natural killer cells induce neutrophil extracellular trap formation in venous thrombosis. J. Thromb. Haemost. 2019, 17, 403–414.
- Chatfield, S.M.; Grebe, K.; Whitehead, L.W.; Rogers, K.L.; Nebl, T.; Murphy, J.M.; Wicks, I.P. Monosodium urate crystals generate nuclease-resistant neutrophil extracellular traps via a distinct molecular pathway. J. Immunol. 2018, 200, 1802–1816.
- Schauer, C.; Janko, C.; Munoz, L.E.; Zhao, Y.; Kienhofer, D.; Frey, B.; Lell, M.; Manger, B.; Rech, J.; Naschberger, E.; et al. Aggregated neutrophil extracellular traps limit inflammation by degrading cytokines and chemokines. Nat. Med. 2014, 20, 511–517.
- Warnatsch, A.; Ioannou, M.; Wang, Q.; Papayannopoulos, V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science 2015, 349, 316–320.
- Menegazzo, L.; Ciciliot, S.; Poncina, N.; Mazzucato, M.; Persano, M.; Bonora, B.; Albiero, M.; Vigili de Kreutzenberg, S.; Avogaro, A.; Fadini, G.P. NETosis is induced by high glucose and associated with type 2 diabetes. Acta Diabetol. 2015, 52, 497–503.
- Wong, S.L.; Demers, M.; Martinod, K.; Gallant, M.; Wang, Y.; Goldfine, A.B.; Kahn, C.R.; Wagner, D.D. Diabetes primes neutrophils to undergo NETosis, which impairs wound healing. Nat. Med. 2015, 21, 815–819.
- Wang, L.; Zhou, X.; Yin, Y.; Mai, Y.; Wang, D.; Zhang, X. Hyperglycemia induces neutrophil extracellular traps formation through an NADPH oxidase-dependent pathway in diabetic retinopathy. Front Immunol. 2018, 9, 3076.
- Hosseinzadeh, A.; Thompson, P.R.; Segal, B.H.; Urban, C.F. Nicotine induces neutrophil extracellular traps. J. Leuko Biol. 2016, 100, 1105–1112.
- Lee, J.; Luria, A.; Rhodes, C.; Raghu, H.; Lingampalli, N.; Sharpe, O.; Rada, B.; Sohn, D.H.; Robinson, W.H.; Sokolove, J. Nicotine drives neutrophil extracellular traps formation and accelerates collagen-induced arthritis. Rheumatology (Oxford) 2017, 56, 644–653.
- Jorch, S.K.; Kubes, P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat. Med. 2017, 23, 279–287.
- Boeltz, S.; Amini, P.; Anders, H.J.; Andrade, F.; Bilyy, R.; Chatfield, S.; Cichon, I.; Clancy, D.M.; Desai, J.; Dumych, T.; et al. To NET or not to NET: current opinions and state of the science regarding the formation of neutrophil extracellular traps. Cell Death Differ. 2019, 26, 395–408.
- Papayannopoulos, V. Neutrophil extracellular traps in immunity and disease. Nat. Rev. Immunol. 2018, 18, 134–147.
- Hakkim, A.; Fuchs, T.A.; Martinez, N.E.; Hess, S.; Prinz, H.; Zychlinsky, A.; Waldmann, H. Activation of the Raf-MEK-ERK pathway is required for neutrophil extracellular trap formation. Nat. Chem. Biol. 2011, 7, 75–77.
- Douda, D.N.; Khan, M.A.; Grasemann, H.; Palaniyar, N. SK3 channel and mitochondrial ROS mediate NADPH oxidase-independent NETosis induced by calcium influx. Proc. Natl. Acad. Sci. USA 2015, 112, 2817–2822.
- Papayannopoulos, V.; Metzler, K.D.; Hakkim, A.; Zychlinsky, A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. J. Cell Biol. 2010, 191, 677–691.
- Erpenbeck, L.; Schon, M.P. Neutrophil extracellular traps: protagonists of cancer progression? Oncogene 2017, 36, 2483–2490.
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665.
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671.
- Chen, K.W.; Monteleone, M.; Boucher, D.; Sollberger, G.; Ramnath, D.; Condon, N.D.; von Pein, J.B.; Broz, P.; Sweet, M.J.; Schroder, K. Noncanonical inflammasome signaling elicits gasdermin D-dependent neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6676.
- Sollberger, G.; Choidas, A.; Burn, G.L.; Habenberger, P.; Di Lucrezia, R.; Kordes, S.; Menninger, S.; Eickhoff, J.; Nussbaumer, P.; Klebl, B.; et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci. Immunol. 2018, 3, eaar6689.
- Thiam, H.R.; Wong, S.L.; Qiu, R.; Kittisopikul, M.; Vahabikashi, A.; Goldman, A.E.; Goldman, R.D.; Wagner, D.D.; Waterman, C.M. NETosis proceeds by cytoskeleton and endomembrane disassembly and PAD4-mediated chromatin decondensation and nuclear envelope rupture. Proc. Natl Acad. Sci. USA 2020, 117, 7326–7337.
- Tatsiy, O.; McDonald, P.P. Physiological stimuli induce PAD4-dependent, ROS-independent NETosis, with early and late events controlled by discrete signaling pathways. Front Immunol. 2018, 9, 2036.
- Maugeri, N.; Campana, L.; Gavina, M.; Covino, C.; De Metrio, M.; Panciroli, C.; Maiuri, L.; Maseri, A.; D’Angelo, A.; Bianchi, M.E.; et al. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J. Thromb. Haemost. 2014, 12, 2074–2088.
- Vogel, S.; Bodenstein, R.; Chen, Q.; Feil, S.; Feil, R.; Rheinlaender, J.; Schaffer, T.E.; Bohn, E.; Frick, J.S.; Borst, O.; et al. Platelet-derived HMGB1 is a critical mediator of thrombosis. J. Clin. Investig. 2015, 125, 4638–4654.
- Nguyen, G.T.; Green, E.R.; Mecsas, J. Neutrophils to the ROScue: Mechanisms of NADPH oxidase activation and bacterial resistance. Front Cell Infect. Microbiol. 2017, 7, 373.
- Chen, K.; Nishi, H.; Travers, R.; Tsuboi, N.; Martinod, K.; Wagner, D.D.; Stan, R.; Croce, K.; Mayadas, T.N. Endocytosis of soluble immune complexes leads to their clearance by FcgammaRIIIB but induces neutrophil extracellular traps via FcgammaRIIA in vivo. Blood 2012, 120, 4421–4431.
- Aleman, O.R.; Mora, N.; Cortes-Vieyra, R.; Uribe-Querol, E.; Rosales, C. Differential use of human neutrophil Fcgamma receptors for inducing neutrophil extracellular trap formation. J. Immunol. Res. 2016, 2016, 2908034.
- Lood, C.; Arve, S.; Ledbetter, J.; Elkon, K.B. TLR7/8 activation in neutrophils impairs immune complex phagocytosis through shedding of FcgRIIA. J. Exp. Med. 2017, 214, 2103–2119.
- Kapoor, S.; Opneja, A.; Nayak, L. The role of neutrophils in thrombosis. Thromb. Res. 2018, 170, 87–96.
- Pertiwi, K.R.; van der Wal, A.C.; Pabittei, D.R.; Mackaaij, C.; van Leeuwen, M.B.; Li, X.; de Boer, O.J. Neutrophil Extracellular Traps Participate in All Different Types of Thrombotic and Haemorrhagic Complications of Coronary Atherosclerosis. Thromb. Haemost. 2018, 118, 1078–1087.
- Mangold, A.; Alias, S.; Scherz, T.; Hofbauer, T.; Jakowitsch, J.; Panzenbock, A.; Simon, D.; Laimer, D.; Bangert, C.; Kammerlander, A.; et al. Coronary Neutrophil Extracellular Trap Burden and Deoxyribonuclease Activity in ST-Elevation Acute Coronary Syndrome Are Predictors of ST-Segment Resolution and Infarct Size. Circ. Res. 2015, 116, 1182–1192.
- Savchenko, A.S.; Martinod, K.; Seidman, M.A.; Wong, S.L.; Borissoff, J.I.; Piazza, G.; Libby, P.; Goldhaber, S.Z.; Mitchell, R.N.; Wagner, D.D. Neutrophil extracellular traps form predominantly during the organizing stage of human venous thromboembolism development. J. Thromb. Haemost. 2014, 12, 860–870.
- Fuchs, T.A.; Brill, A.; Duerschmied, D.; Schatzberg, D.; Monestier, M.; Myers, D.D., Jr.; Wrobleski, S.K.; Wakefield, T.W.; Hartwig, J.H.; Wagner, D.D. Extracellular DNA traps promote thrombosis. Proc. Natl. Acad. Sci. USA 2010, 107, 15880–15885.
- Massberg, S.; Grahl, L.; von Bruehl, M.L.; Manukyan, D.; Pfeiler, S.; Goosmann, C.; Brinkmann, V.; Lorenz, M.; Bidzhekov, K.; Khandagale, A.B.; et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nat. Med. 2010, 16, 887–896.
- Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Thomas, G.M.; Martinod, K.; De Meyer, S.F.; Bhandari, A.A.; Wagner, D.D. Neutrophil extracellular traps promote deep vein thrombosis in mice. J. Thromb. Haemost. 2012, 10, 136–144.
- Quillard, T.; Franck, G.; Mawson, T.; Folco, E.; Libby, P. Mechanisms of erosion of atherosclerotic plaques. Curr. Opin. Lipidol. 2017, 28, 434–441.
- Naruko, T.; Ueda, M.; Haze, K.; van der Wal, A.C.; van der Loos, C.M.; Itoh, A.; Komatsu, R.; Ikura, Y.; Ogami, M.; Shimada, Y.; et al. Neutrophil infiltration of culprit lesions in acute coronary syndromes. Circulation 2002, 106, 2894–2900.
- Megens, R.T.; Vijayan, S.; Lievens, D.; Doring, Y.; van Zandvoort, M.A.; Grommes, J.; Weber, C.; Soehnlein, O. Presence of luminal neutrophil extracellular traps in atherosclerosis. Thromb. Haemost. 2012, 107, 597–598.
- Yunoki, K.; Naruko, T.; Komatsu, R.; Shirai, N.; Nakagawa, M.; Sugioka, K.; Ikura, Y.; Kusano, K.F.; Itoh, A.; Haze, K.; et al. Relation of elevated levels of plasma myeloperoxidase to impaired myocardial microcirculation after reperfusion in patients with acute myocardial infarction. Am. J. Cardiol. 2010, 105, 922–929.
- Libby, P. Mechanisms of acute coronary syndromes and their implications for therapy. N. Engl. J. Med. 2013, 368, 2004–2013.
- Zhao, X.Q.; Dong, L.; Hatsukami, T.; Phan, B.A.; Chu, B.; Moore, A.; Lane, T.; Neradilek, M.B.; Polissar, N.; Monick, D.; et al. MR imaging of carotid plaque composition during lipid-lowering therapy a prospective assessment of effect and time course. Jacc Cardiovasc. Imag. 2011, 4, 977–986.
- Takarada, S.; Imanishi, T.; Kubo, T.; Tanimoto, T.; Kitabata, H.; Nakamura, N.; Tanaka, A.; Mizukoshi, M.; Akasaka, T. Effect of statin therapy on coronary fibrous-cap thickness in patients with acute coronary syndrome: assessment by optical coherence tomography study. Atherosclerosis 2009, 202, 491–497.
- Jernberg, T.; Hasvold, P.; Henriksson, M.; Hjelm, H.; Thuresson, M.; Janzon, M. Cardiovascular risk in post-myocardial infarction patients: nationwide real world data demonstrate the importance of a long-term perspective. Euro. Heart J. 2015, 36, 1163–1170.
- Quillard, T.; Araujo, H.A.; Franck, G.; Shvartz, E.; Sukhova, G.; Libby, P. TLR2 and neutrophils potentiate endothelial stress, apoptosis, and detachment: implications for superficial erosion. Euro. Heart J. 2015, 36, 1394–1404.
- Franck, G.; Mawson, T.; Sausen, G.; Salinas, M.; Masson, G.S.; Cole, A.; Beltrami-Moreira, M.; Chatzizisis, Y.; Quillard, T.; Tesmenitsky, Y.; et al. Flow Perturbation Mediates Neutrophil Recruitment and Potentiates Endothelial Injury via TLR2 in Mice: Implications for Superficial Erosion. Circ. Res. 2017, 121, 31–42.
- Goldmann, B.U.; Rudolph, V.; Rudolph, T.K.; Holle, A.K.; Hillebrandt, M.; Meinertz, T.; Baldus, S. Neutrophil activation precedes myocardial injury in patients with acute myocardial infarction. Free Radic. Biol. Med. 2009, 47, 79–83.
- Borissoff, J.I.; Joosen, I.A.; Versteylen, M.O.; Brill, A.; Fuchs, T.A.; Savchenko, A.S.; Gallant, M.; Martinod, K.; Ten Cate, H.; Hofstra, L.; et al. Elevated levels of circulating DNA and chromatin are independently associated with severe coronary atherosclerosis and a prothrombotic state. Arter. Thromb. Vasc. Biol. 2013, 33, 2032–2040.
- Ferrante, G.; Nakano, M.; Prati, F.; Niccoli, G.; Mallus, M.T.; Ramazzotti, V.; Montone, R.A.; Kolodgie, F.D.; Virmani, R.; Crea, F. High levels of systemic myeloperoxidase are associated with coronary plaque erosion in patients with acute coronary syndromes: a clinicopathological study. Circulation 2010, 122, 2505–2513.
- Barrett, T.J.; Distel, E.; Murphy, A.J.; Hu, J.; Garshick, M.S.; Ogando, Y.; Liu, J.; Vaisar, T.; Heinecke, J.W.; Berger, J.S.; et al. Apolipoprotein AI promotes atherosclerosis regression in diabetic mice by suppressing myelopoiesis and plaque inflammation. Circulation 2019, 140, 1170–1184.
- Qi, H.; Yang, S.; Zhang, L. Neutrophil Extracellular Traps and Endothelial Dysfunction in Atherosclerosis and Thrombosis. Front Immunol. 2017, 8, 928.
- Meegan, J.E.; Yang, X.; Beard, R.S., Jr.; Jannaway, M.; Chatterjee, V.; Taylor-Clark, T.E.; Yuan, S.Y. Citrullinated histone 3 causes endothelial barrier dysfunction. Biochem. Biophys. Res. Commun. 2018, 503, 1498–1502.
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: a predominant role of histones. PLoS ONE 2012, 7, e32366.
- Evrard, S.M.; Lecce, L.; Michelis, K.C.; Nomura-Kitabayashi, A.; Pandey, G.; Purushothaman, K.R.; d’Escamard, V.; Li, J.R.; Hadri, L.; Fujitani, K.; et al. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat. Commun. 2016, 7, 11853.
- Chen, P.Y.; Qin, L.; Baeyens, N.; Li, G.; Afolabi, T.; Budatha, M.; Tellides, G.; Schwartz, M.A.; Simons, M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J. Clin. Inves. 2015, 125, 4514–4528.
- Wesseling, M.; Sakkers, T.R.; de Jager, S.C.A.; Pasterkamp, G.; Goumans, M.J. The morphological and molecular mechanisms of epithelial/endothelial-to-mesenchymal transition and its involvement in atherosclerosis. Vasc. Pharm. 2018, 106, 1–8.
- Pieterse, E.; Rother, N.; Garsen, M.; Hofstra, J.M.; Satchell, S.C.; Hoffmann, M.; Loeven, M.A.; Knaapen, H.K.; van der Heijden, O.W.H.; Berden, J.H.M.; et al. Neutrophil Extracellular Traps Drive Endothelial-to-Mesenchymal Transition. Arterioscler. Thromb. Vasc. Biol. 2017, 37, 1371–1379.
- Drechsler, M.; Megens, R.T.; van Zandvoort, M.; Weber, C.; Soehnlein, O. Hyperlipidemia-triggered neutrophilia promotes early atherosclerosis. Circulation 2010, 122, 1837–1845.
- Doring, Y.; Drechsler, M.; Soehnlein, O.; Weber, C. Neutrophils in atherosclerosis: from mice to man. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 288–295.
- Doring, Y.; Soehnlein, O.; Weber, C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ. Res. 2017, 120, 736–743.
- Kato, R.; Mori, C.; Kitazato, K.; Arata, S.; Obama, T.; Mori, M.; Takahashi, K.; Aiuchi, T.; Takano, T.; Itabe, H. Transient increase in plasma oxidized LDL during the progression of atherosclerosis in apolipoprotein E knockout mice. Arter. Thromb. Vasc. Biol. 2009, 29, 33–39.
- Obama, T.; Ohinata, H.; Takaki, T.; Iwamoto, S.; Sawada, N.; Aiuchi, T.; Kato, R.; Itabe, H. Cooperative action of oxidized low-density lipoproteins and neutrophils on endothelial inflammatory responses through neutrophil extracellular trap formation. Front Immunol. 2019, 10, 1899.
- Jaipersad, A.S.; Lip, G.Y.; Silverman, S.; Shantsila, E. The role of monocytes in angiogenesis and atherosclerosis. J. Am. Coll. Cardiol. 2014, 63, 1–11.
- Cao, W.; Abdelgawwad, M.S.; Li, J.; Zheng, X.L. Apolipoprotein B100/low-density lipoprotein regulates proteolysis and functions of von Willebrand factor under arterial shear. Thromb. Haemost. 2019, 119, 1933–1946.
- Sorvillo, N.; Mizurini, D.M.; Coxon, C.; Martinod, K.; Tilvawala, R.; Cherpokova, D.; Salinger, A.J.; Seward, R.J.; Staudinger, C.; Weerapana, E.; et al. Plasma peptidylarginine deiminase IV promotes VWF-platelet string formation and accelerates thrombosis after vessel injury. Circ. Res. 2019, 125, 507–519.
- Joly, B.S.; Coppo, P.; Veyradier, A. Thrombotic thrombocytopenic purpura. Blood 2017, 129, 2836–2846.
- Smeets, M.W.J.; Mourik, M.J.; Niessen, H.W.M.; Hordijk, P.L. Stasis promotes erythrocyte adhesion to von Willebrand factor. Arter. Thromb. Vasc. Biol. 2017, 37, 1618–1627.


