 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Sylvain Dore | + 1518 word(s) | 1518 | 2021-06-24 04:23:50 | | | |
| 2 | Enzi Gong | Meta information modification | 1518 | 2021-06-25 03:19:29 | | |
Video Upload Options
Hemopexin (Hpx) is a unique internal scavenging glycoprotein that is responsible for the homeostatic maintenance of blood through the regulatory binding of free heme, which eliminates heme’s harmful pro-oxidant and pro-inflammatory potential. Sickle cell disease (SCD) is caused by mutations in the Hb subunit β (HBB) gene coding for the β-globin subunit of the Hb molecule. Specifically, in the series of hemoglobinopathies included under the umbrella term SCD, at least one of the two β-globin subunits of Hb is replaced with the abnormal β-globin hemoglobin S (HbS).
1. Overview
Circulating hemopexin is the primary protein responsible for the clearance of heme; therefore, it is a systemic combatant against deleterious inflammation and oxidative stress induced by the presence of free heme. This role of hemopexin is critical in hemolytic pathophysiology. In this review, we outline the current research regarding how the dynamic activity of hemopexin is implicated in sickle cell disease, which is characterized by a pathological aggregation of red blood cells and excessive hemolysis. This pathophysiology leads to symptoms such as acute kidney injury, vaso-occlusion, ischemic stroke, pain crises, and pulmonary hypertension exacerbated by the presence of free heme and hemoglobin. This review includes in vivo studies in mouse, rat, and guinea pig models of sickle cell disease, as well as studies in human samples. In summary, the current research indicates that hemopexin is likely protective against these symptoms and that rectifying depleted hemopexin in patients with sickle cell disease could improve or prevent the symptoms. The data compiled in this review suggest that further preclinical and clinical research should be conducted to uncover pathways of hemopexin in pathological states to evaluate its potential clinical function as both a biomarker and therapy for sickle cell disease and related hemoglobinopathies.
2. Heme Clearance
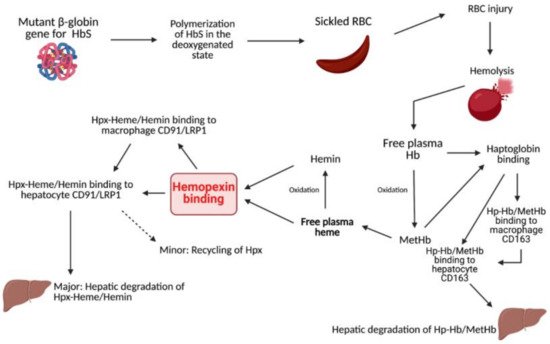
3. Sickle Cell Disease
4. Sickle Cell Disease and Hemolysis
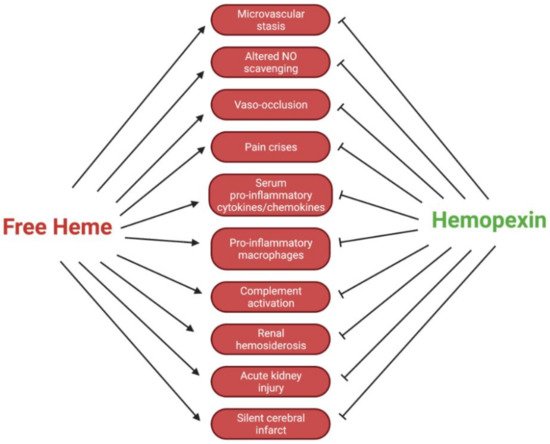
5. Conclusions
References
- Vinchi, F.; Costa da Silva, M.; Ingoglia, G.; Petrillo, S.; Brinkman, N.; Zuercher, A.; Cerwenka, A.; Tolosano, E.; Muckenthaler, M.U. Hemopexin therapy reverts heme-induced pro-inflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 2016, 127, 473–486.
- Cox, M.C.; Le Brun, N.; Thomson, A.J.; Smith, A.; Morgan, W.T.; Moore, G.R. MCD, EPR and NMR Spectroscopic Studies of Rabbit Hemopexin and Its Heme Binding Domain. Biochim. Biophys. Acta 1995, 1253, 215–223.
- Shipulina, N.; Smith, A.; Morgan, W.T. Heme Binding by Hemopexin: Evidence for Multiple Modes of Binding and Functional Implications. J. Protein Chem. 2000, 19, 239–248.
- Detzel, M.S.; Schmalohr, B.F.; Steinbock, F.; Hopp, M.-T.; Ramoji, A.; Paul George, A.A.; Neugebauer, U.; Imhof, D. Revisiting the Interaction of Heme with Hemopexin. Biol. Chem. 2021, 402, 675–691.
- Smith, A.; McCulloh, R.J. Hemopexin and haptoglobin: Allies against heme toxicity from hemoglobin not contenders. Front. Physiol. 2015, 6, 187.
- Belcher, J.D.; Chen, C.; Nguyen, J.; Abdulla, F.; Zhang, P.; Nguyen, H.; Nguyen, P.; Killeen, T.; Miescher, S.M.; Brinkman, N.; et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PLoS ONE 2018, 13, e0196455.
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61.
- Stuart, M.J.; Nagel, R.L. Sickle-cell disease. Lancet 2004, 364, 1343–1360.
- Kato, G.J.; Piel, F.B.; Reid, C.D.; Gaston, M.H.; Ohene-Frempong, K.; Krishnamurti, L.; Smith, W.R.; Panepinto, J.A.; Weatherall, D.J.; Costa, F.F.; et al. Sickle cell disease. Nat. Rev. Dis. Primers 2018, 4, 18010.
- Rees, D.C.; Williams, T.N.; Gladwin, M.T. Sickle-cell disease. Lancet 2010, 376, 2018–2031.
- Aygun, B.; Odame, I. A global perspective on sickle cell disease. Pediatr. Blood Cancer 2012, 59, 386–390.
- Hebbel, R.P.; Morgan, W.T.; Eaton, J.W.; Hedlund, B.E. Accelerated autoxidation and heme loss due to instability of sickle hemoglobin. Proc. Natl. Acad. Sci. USA 1988, 85, 237–241.
- Lei, J.; Paul, J.; Wang, Y.; Gupta, M.; Vang, D.; Thompson, S.; Jha, R.; Nguyen, J.; Valverde, Y.; Lamarre, Y.; et al. Heme Causes Pain in Sickle Mice via Toll-Like Receptor 4-Mediated Reactive Oxygen Species- and Endoplasmic Reticulum Stress-Induced Glial Activation. Antioxid. Redox Signal. 2021, 34, 279–293.
- Figueiredo, R.T.; Fernandez, P.L.; Mourao-Sa, D.S.; Porto, B.N.; Dutra, F.F.; Alves, L.S.; Oliveira, M.F.; Oliveira, P.L.; Graça-Souza, A.V.; Bozza, M.T. Characterization of heme as activator of Toll-like receptor 4. J. Biol. Chem. 2007, 282, 20221–20229.
- Sansing, L.H.; Harris, T.H.; Welsh, F.A.; Kasner, S.E.; Hunter, C.A.; Kariko, K. Toll-like receptor 4 contributes to poor outcome after intracerebral hemorrhage. Ann. Neurol. 2011, 70, 646–656.
- O’Neill, L.A.J.; Golenbock, D.; Bowie, A.G. The history of Toll-like receptors—Redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460.
- Adisa, O.A.; Hu, Y.; Ghosh, S.; Aryee, D.; Osunkwo, I.; Ofori-Acquah, S.F. Association between plasma free haem and incidence of vaso-occlusive episodes and acute chest syndrome in children with sickle cell disease. Br. J. Haematol. 2013, 162, 702–705.
- Sundd, P.; Gladwin, M.T.; Novelli, E.M. Pathophysiology of sickle cell disease. Annu. Rev. Pathol. 2019, 14, 263–292.
- Ansari, J.; Gavins, F.N.E. Ischemia-Reperfusion Injury in Sickle Cell Disease: From Basics to Therapeutics. Am. J. Pathol. 2019, 189, 706–718.
- Li, R.; Saleem, S.; Zhen, G.; Cao, W.; Zhuang, H.; Lee, J.; Smith, A.; Altruda, F.; Tolosano, E.; Doré, S. Heme-hemopexin complex attenuates neuronal cell death and stroke damage. J. Cereb. Blood Flow Metab. 2009, 29, 953–964.
- Dong, B.; Yang, Y.; Zhang, Z.; Xie, K.; Su, L.; Yu, Y. Hemopexin alleviates cognitive dysfunction after focal cerebral ischemia-reperfusion injury in rats. BMC Anesthesiol. 2019, 19, 13.
- Dong, B.; Cai, M.; Fang, Z.; Wei, H.; Zhu, F.; Li, G.; Dong, H.; Xiong, L. Hemopexin induces neuroprotection in the rat subjected to focal cerebral ischemia. BMC Neurosci. 2013, 14, 58.
- Yang, Y.; Dong, B.; Lu, J.; Wang, G.; Yu, Y. Hemopexin reduces blood-brain barrier injury and protects synaptic plasticity in cerebral ischemic rats by promoting EPCs through the HO-1 pathway. Brain Res. 2018, 1699, 177–185.


