 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Yi Ching Esther WAN | + 3482 word(s) | 3482 | 2021-06-21 10:29:29 | | | |
| 2 | Amina Yu | Meta information modification | 3482 | 2021-07-16 10:36:03 | | |
Video Upload Options
Oncohistone mutations refer to clustered mono-allelic missense mutations that often affect only one of the histone genes, the expression of which exhibits oncogenic features. Oncohistones have been an active area of research since the discovery of H3K27M and H3K36M. Recent effort to catalogue missense histone mutations in cancer have uncovered additional oncohistone mutations affecting other histones.
1. Introduction
The nucleosome is the basic repeating unit of the chromatin. While the four types of histones have dissimilar amino acid sequences, their secondary structures can be generalized to a histone fold domain flanked by two disordered tail domains (Figure 1). The tail domains are rich in lysine residues that are subjected to post-translational modifications such as methylation, acetylation, and ubiquitination. Histone modifications are involved in the regulation of a plethora of biological processes such as transcription and DNA damage repair [1].
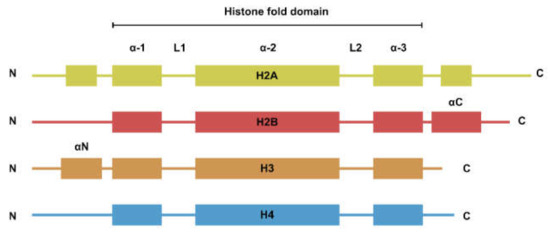
Figure 1. Secondary structure of the four core histones. Histones generally consist of two disordered tails bracketing the histone fold domain and some additional structured fold unique to each type of histone. Helices are represented by rectangles and loops are represented by lines.
Oncohistone mutations are defined as clustered mono-allelic missense mutations that often affect only one of the histone genes (human histones are polygenic in nature; all four histones are encoded by at least fifteen genes), the expression of which exhibits oncogenic features [2][3]. Oncohistones have been an active area of research since 2010, starting with the identification of H3K27M (Histone H3 Lys27-to-Met missense mutation) and H3G34V/R in diffuse intrinsic pontine gliomas [4][5][6], closely followed by the report of H3K36M in chondroblastomas [7] and head and neck squamous cell carcinomas [8][9]. Although these mutations are found in diverse cancer types, they converge functionally to perturb histone post-translational modification and lead to aberrant gene expression. For a detailed review of H3 mutations in cancer, please refer to references [2][3][10][11][12].
Recent efforts to catalogue histone mutations in cancer have vastly expanded the list of potential oncohistones with mechanisms beyond disruption to histone modifications. [13] reported more than 4000 missense mutations occurring in core histone genes, revealing functional convergence of many of the mutations. Some of the most prevalent mutations occur in the globular histone fold domain and are situated in regions important for the structural integrity of the nucleosome, leading to speculation that oncohistones might impede cellular processes beyond histone modifications. This review summarizes recent studies on histone fold mutants occurring in H2B including H2BG53D, H2BE76K/Q, and H2BE113K mutations.
2. H2BG53D Mutation
H2BG53D was identified in 0.07% of all of the cancer patients on cBioPortal. Cancers with H2BG53D mutation include, but are not limited to, pancreatic cancer, glioblastoma, prostate cancer, and lung cancer [14][13]. Long before the identification of H2BG53D in cancer patients, a homologous mutation in fission yeast (htb1-G52D) was reported to cause disruption to gene silencing in the heterochromatic region and defective chromosome segregation [15].
DNA wrapping around the histone octamer is held in place by arginine residues projecting into the minor grooves of nucleosomal DNA [16]. Electrostatic interactions formed around these histone/DNA contact points stabilize the nucleosome and are barriers to processes that require unwinding of DNA. Since glycine-53 of histone H2B (H2BG53) is located in close proximity to one such histone/DNA contact point (H2AR77), the substitution of the neutrally charged glycine to negatively charged aspartate (H2BG53-to-D) was hypothesized to weaken the electrostatic interaction and thus facilitate processes such as nucleosome sliding and transcription.
[17] conducted a restriction enzyme accessibility assay to investigate how H2BG53D affects nucleosome sliding mediated by an ATP-dependent chromatin remodeler. In a restriction enzyme accessibility assay, nucleosomes are reconstituted with DNA containing a restriction enzyme recognition site. In contrast, when the chromatin remodeler is activated by the addition of ATP, the restriction enzyme site will be exposed and cut by restriction enzymes as a result of nucleosome sliding. The restriction enzyme accessibility assay conducted with an H2BG53D-containing nucleosome revealed an elevated remodeling rate, suggesting that H2BG53D might facilitate transcription in cooperation with chromatin remodeling machineries.
The in vitro transcription elongation assay performed by Wan et al. [18] showed that RNA polymerase II progresses quicker on a DNA template containing a H2BG53D nucleosome compared to one that contains a wild-type nucleosome. A single-molecule optical tweezer assay suggests that the increased efficiency by which RNA polymerase II passes through H2BG53D nucleosome is a result of weakened interaction between nucleosomal DNA and the histone octamer.
While H2BG53D’s effect on nucleosome sliding has yet to be examined in a cell-based system, H2BG53D-assocatied transcriptional alterations and its contribution to oncogenesis have been characterized in pancreatic cancer cell lines [18][19].
To study the biological consequence of H2BG53D expression at a physiological level, genome editing mediated by CRISPR/Cas9 was employed to construct stable cell lines that express FLAG-H2B/FLAG-H2BG53D from the endogenousHIST1H2BOlocus. Contrary to the study in yeast where H2BG53D expression inhibits growth [17], H2BG53D expression from its endogenous promoter does not lead to a change in the proliferation of PDAC cell lines. H2BG53D expression in PDAC cell is, however, associated with a gain of migratory properties, indicating changes in gene expression in H2BG53D cell lines. Differentially expressed genes in H2BG53D cell lines are overrepresented in pathways such as the “Rap1-signaling pathway”, “proteoglycans in cancer”, and “ECM-receptor interaction”, all of which are relevant to oncogenesis [19].
Although RNA-seq provided strong evidence for gene expression changes in H2BG53D cell lines, it does not distinguish between transcriptional and post-transcriptional alterations. To investigate whether H2BG53D does indeed alter gene expression on the transcriptional level, Wan and colleagues [19] profiled the nascent transcript level in H2BG53D cell lines by Precision Run- On sequencing (PRO-seq) Consistent with in vitro transcription assay showing increased pol II passaging in H2BG53D nucleosomes, differentially transcribed genes in PRO-seq were dominated by upregulated genes.
The results of RNA-seq and PRO-seq from Wan et al. [19] suggest that H2BG53D expression is associated with elevated transcription. To understand if the transcriptional upregulation is attributable to H2BG53D occupancy, genome-wide distribution of FLAG-H2BG53D was mapped by CUT&RUN [20]. Gene set enrichment analysis revealed that upregulated genes (overlap between PRO-seq and RNA-seq) have higher FLAG enrichment in the FLAG-H2BG53D cell line, indicating the direct contribution of H2BG53D in transcriptional upregulation.
Among the list of upregulated genes with H2BG53D enrichment,ANXA3 was chosen for validating the role of H2BG53D at the gene level. ANXA3 has been implicated in the metastasis of liver cancer [21] and is overexpressed in a number of other cancers. Most importantly, high ANXA3 expression in PDAC patients is correlated with poor overall survival. Together, these findings support a model in which H2BG53D promotes the expression of cancer-associated genes as a result of weakened nucleosomal DNA histone interaction and, subsequently, increased transcription mediated by RNA polymerase II.
2.1 Other histone-DNA contact point mutations
The restriction enzyme accessibility assay performed by Biggert et al. [17] suggested that mutations occurring at histone-DNA contact points, including H2BG53D, converge functionally to enhance nucleosome sliding. Examples of these histone-DNA contact points include H2AR29 and H4R45 (Table 1).
| Residue | Cancer | Post-Translational Modification | Frequencies in Cancer | Oncogenic Mechanism (Proposed) |
|---|---|---|---|---|
| H2BG53 | Pancreas, brain, prostate, lung | No | 0.0007 | Weakens interaction between nucleosomal DNA and increased RNAPII passaging [18], upregulation of cancer and migration-associated genes [19] |
| H2AR29 | Cervix, bladder | Methylation, transcriptional repression [22] | 0.0006 | (Aberrant activation of gene expression) |
| H4R45 | Esophagus, uterus, colon, lung | No | 0.0002 | (Alteration in higher-order chromatin structure, resistance to DNA damages) |
H2AR29 is the second most frequently mutated residue on H2A, identified in 0.06% of patients on corporeal examination. H2AR29P and H2AR29Q accounted for 36% and 56% of all H2AR29 mutations, respectively, and were identified mainly in cervical and bladder cancer [13]. Since the methylation of H2AR29 has been implicated in transcriptional repression [22], it is reasonable to speculate that H2AR29 mutations might lead to aberrant gene activation through the disruption of histone methylation.
H4R45 mutations were identified in 0.02% of cancer patients [13]. H4R45 mutations were initially identified in yeast as a Sin mutation [23] (i.e., switch-independent mutations that alleviate the need for the SWI/SNF remodeling complex in regulating gene expression in yeast). While H4R45C does not alter nucleosomal structure [24], it has been shown to affect Mg2+-dependent higher-order folding of the chromatin array [25]. The expression of H4R45C/H mutants has been associated with enhanced nucleotide excision repair and renders yeast cells more resistant to DNA damage [26].
3. H2BE76 Mutations
H2BE76 is the most frequently mutated H2B residue across all cancer types [14][13]. According to Bennett and colleagues’ analysis [13], polymorphism at E76 is 100 times more common in cancer patients compared to the general population (dbSNP) [27]. Moreover, E76 alterations co-occur with mutations of oncogenes such asRAS, KDM6A, KMT2C, andTP53at frequencies greater than chance events [13].
The E76 residue is buried inside the nucleosome and is situated on the dimer-tetramer interface, suggesting that E76 mutants might contribute to tumorigenesis by nucleosome destabilization. Indeed, both in vitro analysis [14][13][28] and in vivo experiments [28][29] have demonstrated that mutations occurring at the E76 residue would destabilize the H2B–H4 interaction. Although the mechanistic link between nucleosome destabilization and alteration in gene expression remains elusive, the expression of E76 mutants in various cell lines leads to phenotypic change and gene expression profiles distinct from their wild-type counterparts.
H2BE76K accounts for 73.3% of all E76 alterations and was identified in 0.105% of cancer patients on cBioPortal. It was catalogued in a wide array of tumors including, but not limited to, bladder, breast cervical, lung, and ovarian cancer [13].
H2BE76 forms a salt bridge with H4R92 [30] that is disrupted by H2BE76K. Arimura et al. While the glutamate to lysine mutation does not affect the overall backbone geometries of H2A and H3, the side chain of the positively charged lysine causes electrostatic repulsion with H4R92, leading to a drastic conformational change in the α3-helix of H4. This was later corroborated by the molecular dynamic simulation performed by Bennett et al. [13], showing the disruption of the H2B–H4 association by H2BE76K. Since the H2B–H4 interaction is important for the assembly of histone octamer, the disruption of the H2BE76-H4R92 salt bridge is accompanied by nucleosome destabilization.
The instability of the H2BE76K nucleosome as a result of the disrupted H2B–H4 interaction is well supported by in vitro studies. In the absence of DNA, the H2A-H2BE76K dimer fails to be assembled onto the H3-H4 tetramer to form an octamer [13][28]. In the presence of DNA, the reconstitution of the nucleosome with H2BE76K is possible, but the resulting nucleosome releases the H2A-H2B dimer at a lower temperature [28] and is more sensitive to micrococcal nuclease (MNase) digestion compared to its wild-type counterpart [13]. [17] later reported nucleosome instability in both heterotypic and homotypic H2BE76
[17] conducted a high throughput Nap1-mediated histone exchange assay in order to investigate how histone core mutations affect histone exchange rate. In this assay, Nap1 (Nap1 is a histone chaperone responsible for both dimer and tetramer exchange in vivo; Bagert and colleagues have verified that no tetramer exchange occurred under their assay condition) and biotinylated dimers were incubated with a library of uniquely barcoded mono-nucleosomes. More barcoded reads corresponding to the H2BE76K nucleosome were obtained compared to wild-type nucleosome, indicating a higher mobility histone exchange rate in the H2BE76K nucleosome.
The instability and elevated histone exchange of the H2BE76K nucleosome were further confirmed by in vivo experiments. [28] conducted an immunoprecipitation experiment to examine the stoichiometry of the H2BE76K nucleosome in vivo. Moreover, the amount of endogenous H2B pull-down by GFP-H2BE76K was reduced, indicative of nucleosome instability. Bennett and colleagues [13] reported a higher recovery rate of GFP-H2A in MCF10A cells expressing either H2B or H2BE76K, supporting the notion that H2BE76K promotes mobility of H2A-H2B dimer.
Growth defects accompanying H2BE79K expression in yeast [13] (H2BE79 in yeast is homologous to H2BE76 in humans) garnered evidence for the proposition that H2BE76K induces transcriptional alteration. Indeed, RNA-seq performed on both human [13][29] and mouse [17] cell lines showed that H2BE76K cells exhibit a distinct gene expression profile. Colony formation and soft agar assays conducted with various cell lines have uniformly demonstrated the enhanced colony formation ability of H2BE76K cells [13][28][29] (Table 2). [17] reported a differentiation blockade induced by H2BE76K expression in murine mesenchymal progenitor cells, the details of which will be discussed inSection 3.9.
| Cell Type | Assay Condition | RNA-seq | Reference | Remark |
|---|---|---|---|---|
| NIH3T3; Mouse embryonic fibroblast |
Transient expression; colony formation assay |
No | [28] Arimura et al. |
|
| MCF10A; Non-tumorigenic mammary epithelial cells |
Stable expression; soft agar assay |
Yes | [13] Bennett et al. |
H2BE76K enhances colony formation in cooperation with oncogenic PIK3CA-H1047R |
| MDA-MB-231; Breast cancer cells |
CRISPR/Cas9 knockin; colony formation assay |
Yes | [29] Kang et al. |
|
| C3H10T1/2; Mouse mesenchymal progenitors |
Adipocyte and chondrocyte differentiation assay | Yes | [17] Bagert et al. |
Experiments were conducted for both H2BE76K and H2BE76Q cell lines |
The robustness of H2BE76K-induced transcriptional alterations across different cell lines is consistent with the mutation being identified in a wide range of tumor types, suggesting that H2BE76K might be acting through conserved cellular machineries. However, this does not exclude the possibility that H2BE76K might be cooperating with other oncogenes in a tumor type-specific manner.
There is ample evidence supporting the nucleosome destabilization and transcriptional alterations associated with H2BE76K. However, the mechanistic link between the two remains unclear. Current studies paint a picture where H2BE76K shapes the transcriptome by facilitating transcription of its target genes and altering chromatin accessibility.
Bennett and colleagues [13] observed increased sensitivity to MNase in both yeast and mammalian cells expressing H2BE76K, indicating that H2BE76K might alter chromatin accessibility. Chromatin accessibility profiling by ATAC-seq revealed a subset of open chromatin regions that are unique to H2BE76K cells. Intriguingly, 3200 genes, which gained accessibility in their promoter regions, were reported to have higher expression (not to be confused with differentially expressed genes in RNA-seq) in H2BE76K-MCF10A compared to H2B-MCF10A, suggesting that the gain in chromatin accessibility might account for the transcriptional alteration in H2BE76K cells.
While Bennett and colleagues’ work has provided insight into how altered chromatin accessibility in H2BE76K cells is associated with gene expression, the direct effect of H2BE76K on transcription was unknown at the time. To understand the primary consequence of H2BE76K incorporation into the chromatin, Kang and colleagues [29] first profiled the genome-wide distribution H2BE76K to identify regions with H2BE76K enrichment.
CUT&RUN [20] profiling of FLAG K in MDA-MB-231 cells identified more than 2000 genes with significant H2BE76K enrichment. Next, to determine if there is any association between H2BE76K enrichment and transcriptional alteration, upregulated genes in H2BE76K-MDA-MB-231 cells were matched against a gene list ranked by H2BE76K enrichment in gene set enrichment analysis (GSEA). GSEA revealed overrepresentation of the upregulated genes among H2BE76K-enriched genes, which is indicative of the correlation between H2BE76K localization and transcriptional output.
Indeed, further examination of the transcription of ADAM19, an upregulated gene with H2BE76K enrichment, revealed elevated transcriptional activity. Together with the finding that H2BE76K promoted the mobility of the H2A-H2B dimer [31][32], this supports a model in which H2BE76K enhances transcription locally by facilitating the displacement of the H2A-H2B dimer during transcription.
In addition to demonstrating the link between H2BE76K enrichment and transcriptional upregulation in H2BE76K cell lines, profiling of H2BE76K distribution also unveiled specific targeting of the mutant histone into the chromatin. The mechanism underlying the specific targeting of H2BE76 [29] showed that the glutamate to lysine substitution enhances H2B interaction with NAP1L1 and NAP1L2. Although the biological relevance of these phenotypes has not been studied in depth, they suggested that investigation into the interactome of H2BE76K might uncover mechanism governing its deposition.
H2BE76Q accounts for 21.7% of H2BE76 mutations and was found in 0.03% of all patient samples on cBioPortal. Cancers with H2BE76Q mutation include, but are not limited to, lung, breast, cervical, and uterine cancers [13]. Both H2BE76K and H2BE76Q give rise to unstable nucleosomes and lead to similar growth defects when expressed in yeast [13]. However, recent transcriptomic profiling has revealed that the gene expression profile of H2BE76Q-expressing cells is distinct from that of H2BE76K-expressing cells
In vitro studies showed that the stability of the H2BE76Q nucleosome is an intermediate between that of H2BE76K and wild-type nucleosomes. Unlike H2BE76K which fails to form an octamer altogether in the absence of DNA, H2BE76Q is able to form an octamer albeit with lower efficiency compared to wild-type H2B [13]. As reported by Bennett et al. [17], the expression of H2BE76Q or H2BE79Q in yeast leads to growth defects.
H2BE76Q expression in mesenchymal progenitor cells results in transcriptomic alterations distinct from those caused by H2BE76K [17]. Bagert and colleagues [17] performed RNA-seq on mesenchymal progenitor cells stably expressing H2BE76Q and identified more than 2000 differentially expressed genes, 40% of which overlapped with those from H2BE76K-expressing mesenchymal progenitor cells. KEGG pathways unique to H2BE76Q cells include “GAG biosynthesis”, “proteoglycans in cancer”, and “protein digestion”.
Bagert and colleagues [17] speculated that H2BE76 mutants expression might alter cell fate, since the genes regulating stem cells pluripotency are upregulated in both H2BE76K and H2BE76Q cells. To examine the effect of H2BE76 mutants’ expression on cell differentiation, C3H10T1/2 expressing H2BE76K/Q (the same cell lines used for RNA-seq) were subjected to an adipocyte and chondrocyte differentiation assay. C3H10T1/2 is a robust and established model system used to study cell differentiation, the same system that demonstrated differentiation blockade induced by H3K36M [33]. Interestingly, while H2BE76K expression inhibits differentiation into adipocytes and chondrocytes, H2BE76Q expression shows little to no effect.
While both H2BE76K and H2BE76Q lead to nucleosome instability, Bagert and colleagues’ [17] work on H2BE76 mutations suggests that there is an additional mechanism governing the transcriptional alterations associated with H2BE76 mutations. The aforementioned immunoprecipitation performed by Kang et al. [29] demonstrated that H2BE76 K and H2BE76Q affect interactions with different histone chaperones, indicating that the two mutations likely have distinct interactomes which could explain the gene expression changes unique to both mutations.
Other nucleosome-destabilizing histone mutations
H2BE76 mutants represent a class of nucleosome-destabilizing histone mutants. In vitro studies conducted by Bagert et al. [17] have uncovered additional nucleosome-destabilizing mutations which converge on the dimer-tetramer interface, including H2BD68N, H2BE71K/Q, and H4K91N/R.
H2BD68 mutations occurred in 0.05% of cancer patients on cBioPortal, 52% of which result in a D to N substitution identified mainly in lung and uterine cancer [13]. Like H2BE76, H2BD68 also interacts with H4R92 [14]. The expression of H2BD68A/N (or its yeast counterpart H2BD71A) has been shown to prevent growth in yeast [17][34]. In addition, H2BD68A has been shown to impair the binding of H2A.Z-H2B dimers to the chromatin [35].
H2BE71 mutations occurred in 0.06% of cancer patients on cBioPortal, 96% of which led to an E-to-K or E-to-Q substitution. K was identified mainly in breast and skin cancer, whereas H2BE71Q was predominantly found in lung cancer [13]. Transcriptionally, the gene expression profile of H2BE71K-expressing mesenchymal progenitor cells is distinct from that of H2BE71Q-expressing mesenchymal progenitor cells. Phenotypically, only H2BE71K impairs differentiation of mesenchymal progenitor cells into adipocytes.
H4K91 mutations occurred in 0.009% of cancer patients on cBioPortal [13]. H4K91 interacts with H2BE71 to form a salt bridge at the dimer-tetramer interface [14]. Acetylation, mono-ubiquitination, and glutarylation on H4K91 have been shown to regulate chromatin structures and DNA damage response [36][37][38]. Apart from cancer, H4K91 mutations have also been implicated in severe developmental diseases by altering cell cycle control and responses to DNA damage [39][40].
References
- Zhou, K.; Gaullier, G.; Luger, K. Nucleosome structure and dynamics are coming of age. Nat. Struct. Mol. Biol. 2019, 26, 3–13.
- Weinberg, D.N.; Allis, C.D.; Lu, C. Oncogenic mechanisms of histone H3 mutations. Cold Spring Harb. Perspect. Med. 2017, 7, a026443.
- Mohammad, F.; Helin, K. Oncohistones: Drivers of pediatric cancers. Genes Dev. 2017, 31, 2313–2324.
- Schwartzentruber, J.; Korshunov, A.; Liu, X.-Y.; Jones, D.T.; Pfaff, E.; Jacob, K.; Sturm, D.; Fontebasso, A.M.; Quang, D.-A.K.; Tönjes, M.; et al. Driver mutations in histone H3. 3 and chromatin remodelling genes in paediatric glioblastoma. Nature 2012, 482, 226–231.
- Sturm, D.; Witt, H.; Hovestadt, V.; Khuong-Quang, D.-A.; Jones, D.T.; Konermann, C.; Pfaff, E.; Tönjes, M.; Sill, M.; Bender, S.; et al. Hotspot mutations in H3F3A and IDH1 define distinct epigenetic and biological subgroups of glioblastoma. Cancer Cell 2012, 22, 425–437.
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251.
- Behjati, S.; Tarpey, P.S.; Presneau, N.; Scheipl, S.; Pillay, N.; van Loo, P.; Wedge, D.C.; Cooke, S.L.; Gundem, G.; Davies, H.; et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat. Genet. 2013, 45, 1479–1482.
- Shah, M.A.; Denton, E.L.; Arrowsmith, C.H.; Lupien, M.; Schapira, M. A global assessment of cancer genomic alterations in epigenetic mechanisms. Epigenetics Chromatin 2014, 7, 1–15.
- Papillon-Cavanagh, S.; Lu, C.; Gayden, T.; Mikael, L.G.; Bechet, D.; Karamboulas, C.; Ailles, L.; Karamchandani, J.; Marchione, D.M.; Garcia, B.A.; et al. Impaired H3K36 methylation defines a subset of head and neck squamous cell carcinomas. Nat. Genet. 2017, 49, 180–185.
- Wan, Y.C.E.; Liu, J.; Chan, K.M. Histone H3 mutations in cancer. Curr. Pharmacol. Rep. 2018, 4, 292–300.
- Lowe, B.R.; Maxham, L.A.; Hamey, J.J.; Wilkins, M.R.; Partridge, J.F. Histone H3 mutations: An updated view of their role in chromatin deregulation and cancer. Cancers 2019, 11, 660.
- Qiu, L.; Hu, X.; Jing, Q.; Zeng, X.; Chan, K.-M.; Han, J. Mechanism of cancer: Oncohistones in action. J. Genet. Genom. 2018, 45, 227–236.
- Bennett, R.L.; Bele, A.; Small, E.C.; Will, C.M.; Nabet, B.; Oyer, J.A.; Huang, X.; Ghosh, R.P.; Grzybowski, A.T.; Yu, T.; et al. A mutation in histone H2B represents a new class of oncogenic driver. Cancer Discov. 2019, 9, 1438–1451.
- Nacev, B.A.; Feng, L.; Bagert, J.D.; Lemiesz, A.E.; Gao, J.; Soshnev, A.A.; Kundra, R.; Schultz, N.; Muir, T.W.; Allis, C.D. The expanding landscape of ‘oncohistone’mutations in human cancers. Nature 2019, 567, 473–478.
- Maruyama, T.; Nakamura, T.; Hayashi, T.; Yanagida, M. Histone H2B mutations in inner region affect ubiquitination, centromere function, silencing and chromosome segregation. EMBO J. 2006, 25, 2420–2431.
- Cutter, A.R.; Hayes, J.J. A brief review of nucleosome structure. FEBS Lett. 2015, 589, 2914–2922.
- Bagert, J.D.; Mitchener, M.M.; Patriotis, A.L.; Dul, B.E.; Wojcik, F.; Nacev, B.A.; Feng, L.; Allis, C.D.; Muir, T.W. Oncohistone mutations enhance chromatin remodeling and alter cell fates. Nat. Chem. Biol. 2021, 17, 403–4011.
- Wan, Y.C.E.; Leung, T.C.S.; Ding, D.; Sun, X.; Liu, J.; Zhu, L.; Kang, T.Z.E.; Yang, D.; Zhang, Y.; Zhang, J.; et al. Cancer-associated histone mutation H2BG53D disrupts DNA-histone octamer interaction and promotes oncogenic phenotypes. Signal Transduct. Target. Ther. 2020, 5, 1–4.
- Wan, Y.C.E.; Liu, J.; Zhu, L.; Kang, T.Z.E.; Zhu, X.; Lis, J.; Ishibashi, T.; Danko, C.G.; Wang, X.; Chan, K.M. The H2BG53D oncohistone directly upregulates ANXA3 transcription and enhances cell migration in pancreatic ductal adenocarcinoma. Signal Transduct. Target. Ther. 2020, 5, 1–4.
- Meers, M.P.; Bryson, T.D.; Henikoff, J.G.; Henikoff, S. Improved CUT&RUN chromatin profiling tools. Elife 2019, 8, e46314.
- Tong, M.; Fung, T.-M.; Luk, S.T.; Ng, K.-Y.; Lee, T.K.; Lin, C.-H.; Yam, J.W.; Chan, K.W.; Ng, F.; Zheng, B.-J.; et al. ANXA3/JNK signaling promotes self-renewal and tumor growth, and its blockade provides a therapeutic target for hepatocellular carcinoma. Stem Cell Rep. 2015, 5, 45–59.
- Waldmann, T.; Izzo, A.; Kamieniarz, K.; Richter, F.; Vogler, C.; Sarg, B.; Lindner, H.; Young, N.L.; Mittler, G.; Garcia, B.A.; et al. Methylation of H2AR29 is a novel repressive PRMT6 target. Epigenetics Chromatin 2011, 4, 1–10.
- Kruger, W.; Peterson, C.L.; Sil, A.; Coburn, C.; Arents, G.; Moudrianakis, E.N.; Herskowitz, I. Amino acid substitutions in the structured domains of histones H3 and H4 partially relieve the requirement of the yeast SWI/SNF complex for transcription. Genes Dev. 1995, 9, 2770–2779.
- Muthurajan, U.M.; Bao, Y.; Forsberg, L.J.; Edayathumangalam, R.S.; Dyer, P.N.; White, C.L.; Luger, K. Crystal structures of histone Sin. mutant nucleosomes reveal altered protein-DNA interactions. EMBO J. 2004, 23, 260–271.
- Horn, P.J.; Crowley, K.A.; Carruthers, L.M.; Hansen, J.C.; Peterson, C.L. The SIN domain of the histone octamer is essential for intramolecular folding of nucleosomal arrays. Nat. Struct. Biol. 2002, 9, 167–171.
- Nag, R.; Gong, F.; Fahy, D.; Smerdon, M.J. A single amino acid change in histone H4 enhances UV survival and DNA repair in yeast. Nucleic Acids Res. 2008, 36, 3857–3866.
- Sherry, S.T.; Ward, M.-H.; Kholodov, M.; Baker, J.; Phan, L.; Smigielski, E.M.; Sirotkin, K. dbSNP: The NCBI database of genetic variation. Nucleic Acids Res. 2001, 29, 308–311.
- Arimura, Y.; Ikura, M.; Fujita, R.; Noda, M.; Kobayashi, W.; Horikoshi, N.; Sun, J.; Shi, L.; Kusakabe, M.; Harata, M.; et al. Cancer-associated mutations of histones H2B, H3. 1 and H2A. Z. 1 affect the structure and stability of the nucleosome. Nucleic Acids Res. 2018, 46, 10007–10018.
- Kang, T.Z.E.; Zhu, L.; Yang, D.; Ding, D.; Zhu, X.; Wan, Y.C.E.; Liu, J.; Ramakrishnan, S.; Chan, L.L.; Chan, S.Y.; et al. The elevated transcription of ADAM19 by the oncohistone H2BE76K contributes to oncogenic properties in breast cancer. J. Biol. Chem. 2021, 296, 100374.
- Luger, K.; Mäder, A.W.; Richmond, R.K.; Sargent, D.F.; Richmond, T.J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 1997, 389, 251–260.
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703.
- Kujirai, T.; Ehara, H.; Fujino, Y.; Shirouzu, M.; Sekine, S.-i.; Kurumizaka, H. Structural basis of the nucleosome transition during RNA polymerase II passage. Science 2018, 362, 595–598.
- Lu, C.; Jain, S.U.; Hoelper, D.; Bechet, D.; Molden, R.C.; Ran, L.; Murphy, D.; Venneti, S.; Hameed, M.; Pawel, B.R.; et al. Histone H3K36 mutations promote sarcomagenesis through altered histone methylation landscape. Science 2016, 352, 844–849.
- Kawashima, S.; Nakabayashi, Y.; Matsubara, K.; Sano, N.; Enomoto, T.; Tanaka, K.; Seki, M.; Horikoshi, M. Global analysis of core histones reveals nucleosomal surfaces required for chromosome bi-orientation. EMBO J. 2011, 30, 3353–3367.
- Nakabayashi, Y.; Harata, M.; Seki, M. An improved functional analysis of linker-mediated complex (iFALC) strategy. Biochem. Biophys. Res. Commun. 2020, 526, 1164–1169.
- Ye, J.; Ai, X.; Eugeni, E.E.; Zhang, L.; Carpenter, L.R.; Jelinek, M.A.; Freitas, M.A.; Parthun, M.R. Histone H4 lysine 91 acetylation: A core domain modificationassociated with chromatin assembly. Mol. Cell 2005, 18, 123–130.
- Yan, Q.; Dutt, S.; Xu, R.; Graves, K.; Juszczynski, P.; Manis, J.P.; Shipp, M.A. BBAP monoubiquitylates histone H4 at lysine 91 and selectively modulates the DNA damage response. Mol. Cell 2009, 36, 110–120.
- Bao, X.; Liu, Z.; Zhang, W.; Gladysz, K.; Fung, Y.M.E.; Tian, G.; Xiong, Y.; Wong, J.W.H.; Yuen, K.W.Y.; Li, X.D. Glutarylation of histone H4 Lysine 91 regulates chromatin dynamics. Mol. Cell 2019, 76, 660–675.e9.
- Tessadori, F.; Giltay, J.C.; Hurst, J.A.; Massink, M.P.; Duran, K.; Vos, H.R.; van Es, R.M.; Scott, R.H.; van Gassen, K.L.; Bakkers, J. Germline mutations affecting the histone H4 core cause a developmental syndrome by altering DNA damage response and cell cycle control. Nat. Genet. 2017, 49, 1642–1646.
- Tessadori, F.; Rehman, A.U.; Giltay, J.C.; Xia, F.; Streff, H.; Duran, K.; Bakkers, J.; Lalani, S.R.; van Haaften, G. A de novo variant in the human HIST1H4J gene causes a syndrome analogous to the HIST1H4C-associated neurodevelopmental disorder. Eur. J. Hum. Genet. 2020, 28, 674–678.


