 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mariusz J. Nawrocki | + 3442 word(s) | 3442 | 2021-05-12 04:12:28 | | | |
| 2 | Bruce Ren | -21 word(s) | 3421 | 2021-05-24 03:33:09 | | |
Video Upload Options
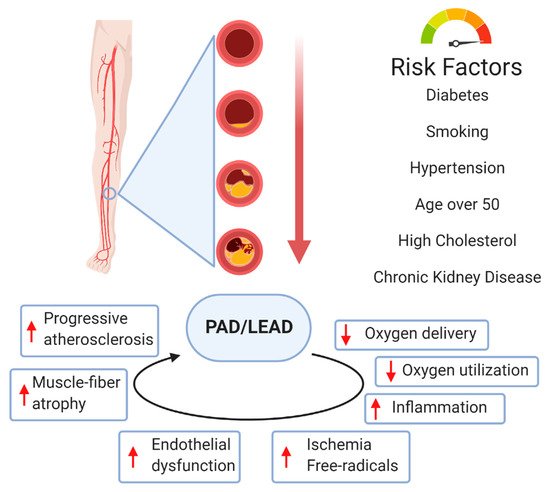
Currently, atherosclerosis, which affects the vascular bed of all vital organs and tissues, is considered as a leading cause of death. Most commonly, atherosclerosis involves coronary and peripheral arteries, which results in acute (e.g., myocardial infarction, lower extremities ischemia) or chronic (persistent ischemia leading to severe heart failure) consequences. All of them have a marked unfavorable impact on the quality of life and are associated with increased mortality and morbidity in human populations. Lower extremity artery disease (LEAD, also defined as peripheral artery disease, PAD) refers to atherosclerotic occlusive disease of the lower extremities, where partial or complete obstruction of peripheral arteries is observed. Decreased perfusion can result in ischemic pain, non-healing wounds, and ischemic ulcers, and significantly reduce the quality of life. However, the progressive atherosclerotic changes cause stimulation of tissue response processes, like vessel wall remodeling and neovascularization. These mechanisms of adapting the vascular network to pathological conditions seem to play a key role in reducing the impact of the changes limiting the flow of blood. Neovascularization as a response to ischemia induces sprouting and expansion of the endothelium to repair and grow the vessels of the circulatory system. Neovascularization consists of three different biological processes: vasculogenesis, angiogenesis, and arteriogenesis. Both molecular and environmental factors that may affect the process of development and growth of blood vessels were analyzed. Particular attention was paid to the changes taking place during LEAD.
1. Introduction
2. Peripheral Arterial Disease
3. Molecular Basis of Vessel Disease
3.1. Neovascularization
3.2. Vasculogenesis
3.3. Angiogenesis
3.4. Arteriogenesis
References
- Bonham, C.A.; Kuehlmann, B.; Gurtner, G.C. Impaired Neovascularization in Aging. Adv. Wound Care 2020, 9, 111–126.
- Carmeliet, P. Mechanisms of angiogenesis and lymphangiogenesis. Nat. Med. 2000, 6, 389–395.
- Helisch, A.; Schaper, W. Arteriogenesis: The development and growth of collateral arteries. Microcirculation 2003, 10, 83–97.
- Rizzi, A.; Benagiano, V.; Ribatti, D. Angiogenesis versus arteriogenesis. Rom. J. Morphol. Embryol. 2017, 58, 15–19.
- Fowkes, F.G.R.; Rudan, D.; Rudan, I.; Aboyans, V.; Denenberg, J.O.; McDermott, M.M.; Norman, P.E.; Sampson, U.K.A.; Williams, L.J.; Mensah, G.A.; et al. Comparison of global estimates of prevalence and risk factors for peripheral artery disease in 2000 and 2010: A systematic review and analysis. Lancet 2013, 382, 1329–1340.
- Sartipy, F.; Sigvant, B.; Lundin, F.; Wahlberg, E. Ten Year Mortality in Different Peripheral Arterial Disease Stages: A Population Based Observational Study on Outcome. Eur. J. Vasc. Endovasc. Surg. 2018, 55, 529–536.
- Aboyans, V.; Ricco, J.B.; Bartelink, M.L.E.L.; Björck, M.; Brodmann, M.; Cohnert, T.; Collet, J.P.; Czerny, M.; De Carlo, M.; Debus, S.; et al. 2017 ESC Guidelines on the Diagnosis and Treatment of Peripheral Arterial Diseases, in collaboration with the European Society for Vascular Surgery (ESVS). Eur. Heart J. 2018, 39, 763–816.
- Norgren, L.; Hiatt, W.R.; Dormandy, J.A.; Nehler, M.R.; Harris, K.A.; Fowkes, F.G.R. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J. Vasc. Surg. 2007, 45, S5–S67.
- De Weerd, M.; Greving, J.P.; De Jong, A.W.F.; Buskens, E.; Bots, M.L. Prevalence of asymptomatic carotid artery stenosis according to age and sex systematic review and metaregression analysis. Stroke 2009, 40, 1105–1113.
- Criqui, M.H.; Aboyans, V. Epidemiology of Peripheral Artery Disease. Circ. Res. 2015, 116, 1509–1526.
- Jude, E.B.; Oyibo, S.O.; Chalmers, N.; Boulton, A.J.M. Peripheral arterial disease in diabetic and nondiabetic patients: A comparison of severity and outcome. Diabetes Care 2001, 24, 1433–1437.
- Aboyans, V.; Criqui, M.H.; Denenberg, J.O.; Knoke, J.D.; Ridker, P.M.; Fronek, A. Risk factors for progression of peripheral arterial disease in large and small vessels. Circulation 2006, 113, 2623–2629.
- Stone, P.A.; Yacoub, M. Inflammatory biomarkers in peripheral arterial disease. Semin. Vasc. Surg. 2014, 27, 148–151.
- Signorelli, S.S.; Marino, E.; Scuto, S.; Di Raimondo, D. Pathophysiology of peripheral arterial disease (Pad): A review on oxidative disorders. Int. J. Mol. Sci. 2020, 21, 4393.
- Kishimoto, Y.; Ibe, S.; Saita, E.; Sasaki, K.; Niki, H.; Miura, K.; Ikegami, Y.; Ohmori, R.; Kondo, K.; Momiyama, Y. Plasma Heme Oxygenase-1 Levels in Patients with Coronary and Peripheral Artery Diseases. Dis. Markers 2018, 2018.
- Walker, M.A.; Hoier, B.; Walker, P.J.; Schulze, K.; Bangsbo, J.; Hellsten, Y.; Askew, C.D. Vasoactive enzymes and blood flow responses to passive and active exercise in peripheral arterial disease. Atherosclerosis 2016, 246, 98–105.
- Xu, J.; Shi, G.P. Vascular wall extracellular matrix proteins and vascular diseases. Biochim. Biophys. Acta Mol. Basis Dis. 2014, 1842, 2106–2119.
- Tanaka, L.Y.; Laurindo, F.R.M. Vascular remodeling: A redox-modulated mechanism of vessel caliber regulation. Free Radic. Biol. Med. 2017, 109, 11–21.
- Hernández-Aguilera, A.; Nielsen, S.H.; Bonache, C.; Fernández-Arroyo, S.; Martín-Paredero, V.; Fibla, M.; Karsdal, M.A.; Genovese, F.; Menendez, J.A.; Camps, J.; et al. Assessment of extracellular matrix-related biomarkers in patients with lower extremity artery disease. J. Vasc. Surg. 2018, 68, 1135–1142.e6.
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of Plaque Formation and Rupture. Circ. Res. 2014, 114, 1852–1866.
- Wight, T.N.; Merrilees, M.J. Proteoglycans in atherosclerosis and restenosis: Key roles for versican. Circ. Res. 2004, 94, 1158–1167.
- Vassiliadis, E.; Barascuk, N.; Karsdal, M.A. Atherofibrosis—A unique and common process of the disease pathogenesis of atherosclerosis and fibrosis—Lessons for biomarker development. Am. J. Transl. Res. 2013, 5, 1–14.
- Blankenberg, S.; Rupprecht, H.J.; Poirier, O.; Bickel, C.; Smieja, M.; Hafner, G.; Meyer, J.; Cambien, F.; Tiret, L. Plasma concentrations and genetic variation of matrix metalloproteinase 9 and prognosis of patients with cardiovascular disease. Circulation 2003, 107, 1579–1585.
- Yan, H.; Chang, Z.; Liu, Z. The risk factors for calcification vary among the different sections of the lower extremity artery in patients with symptomatic peripheral arterial disease. BMC Cardiovasc. Disord. 2020, 20.
- Demer, L.L.; Tintut, Y. Inflammatory, metabolic, and genetic mechanisms of vascular calcification. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 715–723.
- Chistiakov, D.A.; Myasoedova, V.A.; Melnichenko, A.A.; Grechko, A.V.; Orekhov, A.N. Calcifying Matrix Vesicles and Atherosclerosis. BioMed Res. Int. 2017, 2017.
- Wang, Y.; Chen, J.; Zhang, Y.; Yu, W.; Zhang, C.; Gong, L.; Shao, L.; Lu, J.; Gao, Y.; Chen, X.; et al. Common genetic variants of MGP are associated with calcification on the arterial wall but not with calcification present in the atherosclerotic plaques. Circ. Cardiovasc. Genet. 2013, 6, 271–278.
- Lanzer, P.; Boehm, M.; Sorribas, V.; Thiriet, M.; Janzen, J.; Zeller, T.; St Hilaire, C.; Shanahan, C. Medial vascular calcification revisited: Review and perspectives. Eur. Heart J. 2014, 35, 1515–1525.
- Kullo, I.J.; Leeper, N.J. The Genetic Basis of Peripheral Arterial Disease: Current Knowledge, Challenges, and Future Directions. Circ. Res. 2015, 116, 1551–1560.
- Flex, A.; Gaetani, E.; Pola, R.; Santoliquido, A.; Aloi, F.; Papaleo, P.; Dal Lago, A.; Pola, E.; Serricchio, M.; Tondi, P.; et al. The -174 G/C polymorphism of the interleukin-6 gene promoter is associated with peripheral artery occlusive disease. Eur. J. Vasc. Endovasc. Surg. 2002, 24, 264–268.
- Li, R.; Folsom, A.R.; Sharrett, A.R.; Couper, D.; Bray, M.; Tyroler, H.A. Interaction of the glutathione S-transferase genes and cigarette smoking on risk of lower extremity arterial disease: The Atherosclerosis Risk in Communities (ARIC) study. Atherosclerosis 2001, 154, 729–738.
- Arellano Mendoza, M.; Robles, H.; Romo, E.; Rios, A.; Escalante, B. Nitric Oxide-Dependent Neovascularization Role in the Lower Extremity Disease. Curr. Pharm. Des. 2007, 13, 3591–3596.
- Vogel, J.; Niederer, D.; Jung, G.; Troidl, K. Exercise-Induced Vascular Adaptations under Artificially Versus Pathologically Reduced Blood Flow: A Focus Review with Special Emphasis on Arteriogenesis. Cells 2020, 9, 333.
- Chalothorn, D.; Faber, J.E. Formation and maturation of the native cerebral collateral circulation. J. Mol. Cell. Cardiol. 2010, 49, 251–259.
- Ribatti, D.; Vacca, A.; Nico, B.; Roncali, L.; Dammacco, F. Postnatal vasculogenesis. Mech. Dev. 2001, 100, 157–163.
- Simons, M.; Eichmann, A. Molecular controls of arterial morphogenesis. Circ. Res. 2015, 116, 1712–1724.
- Pugh, C.W.; Ratcliffe, P.J. Regulation of angiogenesis by hypoxia: Role of the HIF system. Nat. Med. 2003, 9, 677–684.
- Stetler-Stevenson, W.G. Matrix metalloproteinases in angiogenesis: A moving target for therapeutic intervention. J. Clin. Investig. 1999, 103, 1237–1241.
- Faber, J.E.; Chilian, W.M.; Deindl, E.; Van Royen, N.; Simons, M. A brief etymology of the collateral circulation. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 1854–1859.
- Lasch, M.; Caballero Martinez, A.; Kumaraswami, K.; Ishikawa-Ankerhold, H.; Meister, S.; Deindl, E. Contribution of the Potassium Channels KV1.3 and KCa3.1 to Smooth Muscle Cell Proliferation in Growing Collateral Arteries. Cells 2020, 9, 913.
- Ricard, N.; Zhang, J.; Zhuang, Z.W.; Simons, M. Isoform-Specific Roles of ERK1 and ERK2 in Arteriogenesis. Cells 2019, 9, 38.
- Sealock, R.; Zhang, H.; Lucitti, J.L.; Moore, S.M.; Faber, J.E. Congenic fine-mapping identifies a major causal locus for variation in the native collateral circulation and ischemic injury in brain and lower extremity. Circ. Res. 2014, 114, 660–671.
- Peirce, S.M.; Skalak, T.C. Microvascular remodeling: A complex continuum spanning angiogenesis to arteriogenesis. Microcirculation 2003, 10, 99–111.
- Troidl, K.; Tribulova, S.; Cai, W.J.; Rüding, I.; Apfelbeck, H.; Schierling, W.; Troidl, C.; Schmitz-Rixen, T.; Schaper, W. Effects of endogenous nitric oxide and of deta nonoate in arteriogenesis. J. Cardiovasc. Pharmacol. 2010, 55, 153–160.
- Buschmann, I.; Pries, A.; Styp-Rekowska, B.; Hillmeister, P.; Loufrani, L.; Henrion, D.; Shi, Y.; Duelsner, A.; Hoefer, I.; Gatzke, N.; et al. Pulsatile shear and Gja5 modulate arterial identity and remodeling events during flow-driven arteriogenesis. Development 2010, 137, 2187–2196.
- Tronc, F.; Mallat, Z.; Lehoux, S.; Wassef, M.; Esposito, B.; Tedgui, A. Role of Matrix Metalloproteinases in Blood Flow–Induced Arterial Enlargement. Arterioscler. Thromb. Vasc. Biol. 2000, 20.
- Shi, Z.D.; Tarbell, J.M. Fluid flow mechanotransduction in vascular smooth muscle cells and fibroblasts. Ann. Biomed. Eng. 2011, 39, 1608–1619.
- Gerhold, K.A.; Schwartz, M.A. Ion channels in endothelial responses to fluid shear stress. Physiology 2016, 31, 359–369.
- Sayed, A.; Schierling, W.; Troidl, K.; Rüding, I.; Nelson, K.; Apfelbeck, H.; Benli, I.; Schaper, W.; Schmitz-Rixen, T. Exercise linked to transient increase in expression and activity of cation channels in newly formed hind-limb collaterals. Eur. J. Vasc. Endovasc. Surg. 2010, 40, 81–87.
- Troidl, C.; Troidl, K.; Schierling, W.; Cai, W.J.; Nef, H.; Möllmann, H.; Kostin, S.; Schimanski, S.; Hammer, L.; Elsässer, A.; et al. Trpv4 induces collateral vessel growth during regeneration of the arterial circulation. J. Cell. Mol. Med. 2009, 13, 2613–2621.
- Pfenniger, A.; Wong, C.; Sutter, E.; Cuhlmann, S.; Dunoyer-Geindre, S.; Mach, F.; Horrevoets, A.J.; Evans, P.C.; Krams, R.; Kwak, B.R. Shear stress modulates the expression of the atheroprotective protein Cx37 in endothelial cells. J. Mol. Cell. Cardiol. 2012, 53, 299–309.
- Chen, Z.; Rubin, J.; Tzima, E. Role of PECAM-1 in arteriogenesis and specification of preexisting collaterals. Circ. Res. 2010, 107, 1355–1363.
- Takeda, Y.; Costa, S.; Delamarre, E.; Roncal, C.; Leite De Oliveira, R.; Squadrito, M.L.; Finisguerra, V.; Deschoemaeker, S.; Bruyère, F.; Wenes, M.; et al. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 2011, 479, 122–126.
- de Paula, E.V.; Flores-Nascimento, M.C.; Arruda, V.R.; Garcia, R.A.; Ramos, C.D.; Guillaumon, A.T.; Annichino-Bizzacchi, J.M. Dual gene transfer of fibroblast growth factor-2 and platelet derived growth factor-BB using plasmid deoxyribonucleic acid promotes effective angiogenesis and arteriogenesis in a rodent model of hindlimb ischemia. Transl. Res. 2009, 153, 232–239.
- Troidl, K.; Schubert, C.; Vlacil, A.K.; Chennupati, R.; Koch, S.; Schütt, J.; Oberoi, R.; Schaper, W.; Schmitz-Rixen, T.; Schieffer, B.; et al. The Lipopeptide MALP-2 Promotes Collateral Growth. Cells 2020, 9, 997.
- Deindl, E.; Buschmann, I.; Hoefer, I.E.; Podzuweit, T.; Boengler, K.; Vogel, S.; Van Royen, N.; Fernandez, B.; Schaper, W. Role of ischemia and of hypoxia-inducible genes in arteriogenesis after femoral artery occlusion in the rabbit. Circ. Res. 2001, 89, 779–786.
- Moraes, F.; Paye, J.; Gabhann, F.M.; Zhuang, Z.W.; Zhang, J.; Lanahan, A.A.; Simons, M. Endothelial cell-dependent regulation of arteriogenesis. Circ. Res. 2013, 113, 1076–1086.
- Lanahan, A.A.; Lech, D.; Dubrac, A.; Zhang, J.; Zhuang, Z.W.; Eichmann, A.; Simons, M. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation 2014, 130, 902–909.
- Lanahan, A.; Zhang, X.; Fantin, A.; Zhuang, Z.; Rivera-Molina, F.; Speichinger, K.; Prahst, C.; Zhang, J.; Wang, Y.; Davis, G.; et al. The Neuropilin 1 Cytoplasmic Domain Is Required for VEGF-A-Dependent Arteriogenesis. Dev. Cell 2013, 25, 156–168.
- Van Royen, N.; Piek, J.J.; Buschmann, I.; Hoefer, I.; Voskuil, M.; Schaper, W. Stimulation of arteriogenesis; a new concept for the treatment of arterial occlusive disease. Cardiovasc. Res. 2001, 49, 543–553.
- Shireman, P.K. The chemokine system in arteriogenesis and hind limb ischemia. J. Vasc. Surg. 2007, 45, A48–A56.
- Stabile, E.; Kinnaird, T.; La Sala, A.; Hanson, S.K.; Watkins, C.; Campia, U.; Shou, M.; Zbinden, S.; Fuchs, S.; Kornfeld, H.; et al. CD8+ T lymphocytes regulate the arteriogenic response to ischemia by infiltrating the site of collateral vessel development and recruiting CD4+ mononuclear cells through the expression of interleukin-16. Circulation 2006, 113, 118–124.
- Krishnasamy, K.; Limbourg, A.; Kapanadze, T.; Gamrekelashvili, J.; Beger, C.; Häger, C.; Lozanovski, V.J.; Falk, C.S.; Napp, L.C.; Bauersachs, J.; et al. Blood vessel control of macrophage maturation promotes arteriogenesis in ischemia. Nat. Commun. 2017, 8, 1–14.
- Ribatti, D. A new role of mast cells in arteriogenesis. Microvasc. Res. 2018, 118, 57–60.
- Heil, M.; Ziegelhoeffer, T.; Pipp, F.; Kostin, S.; Martin, S.; Clauss, M.; Schaper, W. Blood monocyte concentration is critical for enhancement of collateral artery growth. Am. J. Physiol. Hear. Circ. Physiol. 2002, 283.
- Morrison, A.R.; Yarovinsky, T.O.; Young, B.D.; Moraes, F.; Ross, T.D.; Ceneri, N.; Zhang, J.; Zhuang, Z.W.; Sinusas, A.J.; Pardi, R.; et al. Chemokine-coupled β2 integrin-induced macrophage Rac2-myosin IIA interaction regulates VEGF-A mRNA stability and arteriogenesis. J. Exp. Med. 2014, 211, 1957–1968.
- Chillo, O.; Kleinert, E.C.; Lautz, T.; Lasch, M.; Pagel, J.I.; Heun, Y.; Troidl, K.; Fischer, S.; Caballero-Martinez, A.; Mauer, A.; et al. Perivascular Mast Cells Govern Shear Stress-Induced Arteriogenesis by Orchestrating Leukocyte Function. Cell Rep. 2016, 16, 2197–2207.
- Bot, I.; van der Velden, D.; Bouwman, M.; Kröner, M.J.; Kuiper, J.; Quax, P.H.A.; de Vries, M.R. Local Mast Cell Activation Promotes Neovascularization. Cells 2020, 9, 701.
- Dopheide, J.F.; Rubrech, J.; Trumpp, A.; Geissler, P.; Zeller, G.C.; Schnorbus, B.; Schmidt, F.; Gori, T.; Münzel, T.; Espinola-Klein, C. Supervised exercise training in peripheral arterial disease increases vascular shear stress and profunda femoral artery diameter. Eur. J. Prev. Cardiol. 2017, 24, 178–191.
- Pope, Z.K.; Willardson, J.M.; Schoenfeld, B.J. Exercise and blood flow restriction. J. Strength Cond. Res. 2013, 27, 2914–2926.
- Loenneke, J.P.; Wilson, G.J.; Wilson, J.M. A mechanistic approach to blood flow occlusion. Int. J. Sports Med. 2010, 31, 1–4.
- Casey, D.P.; Madery, B.D.; Curry, T.B.; Eisenach, J.H.; Wilkins, B.W.; Joyner, M.J. Nitric oxide contributes to the augmented vasodilatation during hypoxic exercise. J. Physiol. 2010, 588, 373–385.


