 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Kenichi Kashimada | + 1018 word(s) | 1018 | 2021-08-10 12:20:41 | | | |
| 2 | Catherine Yang | Meta information modification | 1018 | 2021-08-30 05:08:45 | | | | |
| 3 | Catherine Yang | Meta information modification | 1018 | 2021-08-30 05:09:41 | | |
Video Upload Options
Congenital adrenal hyperplasia (CAH) is an inherited disorder caused by the absence or severely impaired activity of steroidogenic enzymes involved in cortisol biosynthesis. More than 90% of cases result from 21-hydroxylase deficiency (21OHD). To prevent life-threatening adrenal crisis and to help perform appropriate sex assignments for affected female patients, newborn screening (NBS) programs for the classical form of CAH have been introduced in numerous countries.
1. Introduction
Congenital adrenal hyperplasia (CAH) is an inherited disorder caused by the loss or severely impaired activity of steroidogenic enzymes involved in cortisol biosynthesis ( Figure 1 A,B) [1][2]. More than 90 percent of cases result from 21-hydroxylase deficiency (21OHD) caused by mutations in CYP21A2 . The prevalence of 21OHD is estimated to be 1:15,000–16,000 in the USA and Europe [3] and slightly lower in Japan (1:18,000) [4][5][6]. The clinical spectrum of the disease ranges from the most severe to mild forms, depending upon the degree of enzyme deficiency [2].
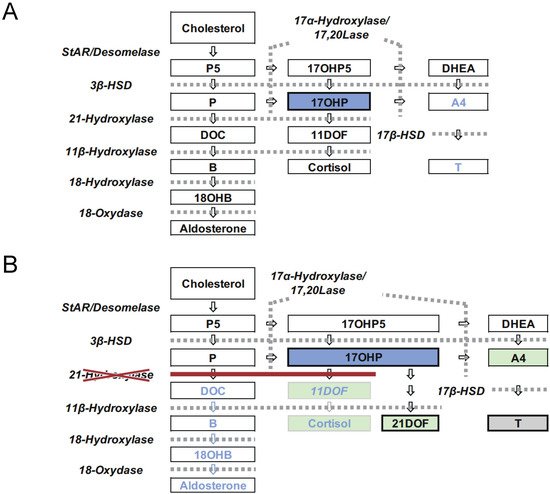
The disease is mainly classified into two forms: classical and nonclassical. The classical form is associated with two major problems: life-threatening adrenal crisis in both sexes and virilization of the external genitalia in 46,XX patients. The classical form is further subdivided into two subtypes, the severest, salt wasting (SW) form, and simple virilizing (SV) form. The SW form is associated with cortisol and aldosterone deficiencies, in which neonates are likely to develop life-threatening adrenal crises with severe hyponatremia and hyperkalemia. Virilization of the external genitalia in newborn females and precocious puberty due to overproduction of androgens by the adrenal cortex are the other major clinical manifestations of the SW and SV forms [2].
To prevent a life-threatening adrenal crisis and help perform appropriate sex assignments for affected female patients, newborn screening (NBS) programs for the classical form of CAH have been introduced in numerous countries [7]. In Japan, the NBS for CAH was introduced in 1989, following that for PKU and congenital hypothyroidism [4].
2. Screening System in Japan
The NBS has two different cutoff values: for “screening positive” and for “retest”. When the 17αOHP level is higher than the screening positive cutoff value, the neonate is directly referred to a pediatric endocrinologist for further endocrinological evaluation. Neonates with 17αOHP levels more than the retest cutoff value are retested. When the 17αOHP levels are higher than the retest cutoff value two–three times, the screening is considered positive ( Figure 2 ) [4][5][8][9][10].
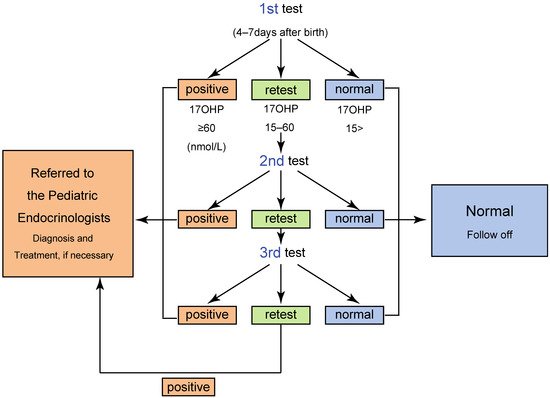
One of the most serious issues in the screening for 21OHD is false positive in preterm neonates. For reducing false positive, some laboratories, including that of Tokyo, employ cutoff values based on gestational age and/or birth weight.The algorithm and criteria for the screening in Tokyo are shown in Table 1 and Figure 2 [5], respectively. By employing the system, a total of 410 infants were judged positive from 1989 to 2013, and of these, 106 patients were diagnosed with CAH, indicating a positive predictive value (PPV) of 25.8 % [5].
Table 1. Criteria of CAH mass screening in Tokyo.
| <Criteria According to the Gestational Age> | |||||
|---|---|---|---|---|---|
| Gestational age at birth (weeks) *1a | ≤29 | 30–34 | 35–36 | ≥37 | |
| Corrected gestational age (weeks) *1b | ≤31 | 32–35 | 36–37 | ≥38 | |
| <Criteria According to Weight> *2,*3 | |||||
| Body weight (g) | ≤999 | 1000–1999 | 2000–2499 | ≥2500 | |
| Cutoff level of 17αOHP [n·mol/L] | Retest | 60 | 45 | 24 | 15 |
| Positive | 60 | 60 | 60 | ||
*1a Samples collected before the age of 7 days, *1b Samples collected at the age of 7 days or after, *2 1st test: body weight = birth weight, 2nd test and after: body weight = corrected body weight calculated by the formula as below. Corrected body weight at test (g) = birth weight (g) + (age at test − 7) × 20 (g). *3 For infants born small or large for gestational age, either the criteria of gestational age (corrected gestational age) or body weight was applied, whichever was a lower value [10].
3. Clinical Outcomes of the Newborn Screening for CAH in Japan
After the introduction of the screening, the clinical outcomes of 21OHD during the neonatal/infantile period were remarkably improved. The average ages at the first visit were 8.2 and 7.6 days (male: 9.2 days, female: 6.0 days) in Sapporo and Tokyo, respectively (Table 2) [6][10]. No fatal cases have been identified. Further, based on a survey for the literature and the annual reports from NBS programs, no false-negative cases have been reported since the introduction of the screening [5][6][10]. We presume that the sex of all 46,XX cases was correctly assigned.
Eradicating the lethal cases of 21OHD is one of the major goals of newborn screening [11]. Despite no reported fatal cases, 37.4% of 21OHD neonates already developed severe salt wasting, which is defined by Na < 130 mEq/L, K > 7 mEq/L, on arrival at medical hospitals in Tokyo screening [10].
Cosistently, in classical 21OHD patients, the serum Na and K levels linearly deteriorated with age in days, and the age when the regression lines reached Na < 130 mEq/L, K > 7 mEq/L approximately coincided at 11.1 and 12.3 days, respectively ( Figure 3 ).
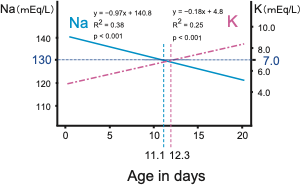
Figure 3. Temporal changes of serum sodium (Na) and potassium (K) levels in 21OHD neonates. Modified the figure from Gau et al, 2020 [10]
4. Database Composition and Improvement of Screening Program
For better and more efficient management of the CAH, the screening programs need persistent improvement in quality. By examining reliable follow-up studies, the outcomes and the experiences of the screening should be retrospectively evaluated and shared among the screening laboratories [12][13][14]. For short-term outcomes, most laboratories and local governments have introduced follow-up surveys in Japan, using the results for more efficient screening by decreasing false positives and early availability of screening results. On the other hand, the assessment of long-term outcomes for CAH patients identified by screening is challenging. A nationwide registry system is required to establish efficient long-term follow-up systems.
Ideally, for establishing feedback systems with long-term follow-up surveys, close collaboration among the screening laboratory professionals, pediatricians, primary care providers, and clinical epidemiologists is essential [13][14][15][16]. A nationwide registry system in which all results of infants are registered and evaluated periodically would lead to further methodological improvements.
References
- Speiser, P.W.; White, P.C. Congenital adrenal hyperplasia. N. Engl. J. Med. 2003, 349, 776–788.
- White, P.C.; Speiser, P.W. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency. Endocr. Rev. 2000, 21, 245–291.
- van der Kamp, H.J.; Wit, J.M. Neonatal screening for congenital adrenal hyperplasia. Eur. J. Endocrinol. 2004, 151 (Suppl. S3), U71–U75.
- Suwa, S. Nationwide survey of neonatal mass-screening for congenital adrenal hyperplasia in Japan. Screening 1994, 3, 141–151.
- Tsuji, A.; Konishi, K.; Hasegawa, S.; Anazawa, A.; Onishi, T.; Ono, M.; Morio, T.; Kitagawa, T.; Kashimada, K. Newborn screening for congenital adrenal hyperplasia in Tokyo, Japan from 1989 to 2013: A retrospective population-based study. BMC Pediatr. 2015, 15, 209.
- Morikawa, S.; Nakamura, A.; Fujikura, K.; Fukushi, M.; Hotsubo, T.; Miyata, J.; Ishizu, K.; Tajima, T. Results from 28 years of newborn screening for congenital adrenal hyperplasia in sapporo. Clin. Pediatr. Endocrinol. 2014, 23, 35–43.
- Clayton, P.E.; Miller, W.L.; Oberfield, S.E.; Ritzen, E.M.; Sippell, W.G.; Speiser, P.W.; Group, E.L.C.W. Consensus statement on 21-hydroxylase deficiency from the European Society for Paediatric Endocrinology and the Lawson Wilkins Pediatric Endocrine Society. Horm. Res. 2002, 58, 188–195.
- Tajima, T.; Fujikura, K.; Fukushi, M.; Hotsubo, T.; Mitsuhashi, Y. Neonatal screening for congenital adrenal hyperplasia in Japan. Pediatr. Endocrinol. Rev. 2012, 10 (Suppl. S1), 72–78.
- Tajima, T.; Fukushi, M. Neonatal mass screening for 21-hydroxylase deficiency. Clin. Pediatr. Endocrinol. 2016, 25, 1–8.
- Gau, M.; Konishi, K.; Takasawa, K.; Nakagawa, R.; Tsuji-Hosokawa, A.; Hashimoto, A.; Sutani, A.; Tajima, T.; Hasegawa, T.; Morio, T.; et al. The progression of salt wasting and the body weight change during the first two weeks of life in classical 21-hydroxylase deficiency patients. Clin. Endocrinol. 2020, 94, 229–236.
- Speiser, P.W.; Arlt, W.; Auchus, R.J.; Baskin, L.S.; Conway, G.S.; Merke, D.P.; Meyer-Bahlburg, H.F.L.; Miller, W.L.; Murad, M.H.; Oberfield, S.E.; et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase deficiency: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2018, 103, 4043–4088.
- Loeber, J.G.; Platis, D.; Zetterstrom, R.H.; Almashanu, S.; Boemer, F.; Bonham, J.R.; Borde, P.; Brincat, I.; Cheillan, D.; Dekkers, E.; et al. Neonatal screening in Europe revisited: An ISNS perspective on the current state and developments since 2010. Int. J. Neonatal. Screen 2021, 7, 15.
- Burgard, P.; Rupp, K.; Lindner, M.; Haege, G.; Rigter, T.; Weinreich, S.S.; Loeber, J.G.; Taruscio, D.; Vittozzi, L.; Cornel, M.C.; et al. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 2. From screening laboratory results to treatment, follow-up and quality assurance. J. Inherit. Metab. Dis. 2012, 35, 613–625.
- Advisory Committee on Heritable Disorders in Newborns and Children; Zuckerman, A.E.; Badawi, D.; Brosco, J.P.; Brower, A.; Eichwald, J.; Feuchtbaum, L.; Finitzo, T.; Flannery, D.; Green, N.; et al. The Role of Quality Measures to Promote Long-Term Follow-up of Children Identified by Newborn Screening Programs; Advisory Committee on Heritable Disorders in Newborns and Children Advisory Committee on Heritable Disorders in Newborns and Children. 2018. Available online: https://www.hrsa.gov/sites/default/files/hrsa/advisory-committees/heritable-disorders/reports-recommendations/reports/role-of-quality-measures-in-nbs-sept2018-508c.pdf” (accessed on 23 April 2021).
- Hoff, T.; Hoyt, A.; Therrell, B.; Ayoob, M. Exploring barriers to long-term follow-up in newborn screening programs. Genet. Med. 2006, 8, 563–570.
- Loeber, J.G.; Burgard, P.; Cornel, M.C.; Rigter, T.; Weinreich, S.S.; Rupp, K.; Hoffmann, G.F.; Vittozzi, L. Newborn screening programmes in Europe; arguments and efforts regarding harmonization. Part 1. From blood spot to screening result. J. Inherit. Metab. Dis. 2012, 35, 603–611.


