 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Antonio Mario Scanu | + 2092 word(s) | 2092 | 2021-02-17 04:49:40 | | | |
| 2 | Vicky Zhou | Meta information modification | 2092 | 2021-02-17 09:09:18 | | |
Video Upload Options
Colorectal cancer stem cells (CCSCs) is a small cells population with stemness behaviors and responsible for tumor progression, recurrence, and therapy resistance. The generation of CCSCs is probably connected to genetic changes in members of signaling pathways, which control self-renewal and pluripotency in SCs and then establish function and phenotype of CCSCs. Particularly, various deregulated CCSC-related miRNAs have been reported to modulate stemness features, controlling CCSCs functions such as regulation of cell cycle genes expression, epithelial-mesenchymal transition, metastasization, and drug-resistance mechanisms. Primarily, CCSC-related miRNAs work by regulating mainly signal pathways known to be involved in CCSCs biology.
1. Introduction
Colorectal cancer (CRC) is the third most common cancer and the second most frequent source of cancer-related mortality worldwide [1]. Arnold et al. recognized three distinct global temporal patterns to CRC development trends: A rise in both incidence and mortality (Baltic countries, Russia, China, and Brazil); an increasing incidence, but decreasing in mortality (Canada, the United Kingdom, Denmark, and Singapore); and reduction in both incidence and mortality (the United States, Japan, and France) [2]. In highly advanced nations, the increased cancer-incidence highlights the influence of dietary habits, obesity, and lifestyle. The reduced cancer-mortality is attributable to population-based screenings and significant progresses in therapeutic options improving CRC patient’s management [2]. Approximately 10% of CRC patients under 55 years showed more severe and unfavorable pathological features than older cohorts, resulting in a negative impact on their survival outcome [3].
CRC is a well-studied malignancy for which extensive and heterogeneous genomic aberrations, well-defined risk factors, slow progression, and identifiable and treatable preneoplastic lesions have been described [4][5]. In patients at stage I of the disease, the five-year survival rate is 90%, but we observed that a drastic reduction of slightly more than 10% is observed when cancer patients reach stage IV [6]. Approximately 20% of CRC patients already have metastases at diagnosis, and metastatic CRC (mCRC) is generally an incurable disease [7]. CRC is a heterogeneous multifactorial disease presenting significant differences in prognoses and responses to treatment. The importance of detecting specific pathway abnormalities is crucial to improve diagnosis, prognosis, and therapeutic strategies. CRC molecular alterations permit us to identify two distinct genetic pathways. The adenoma-carcinoma pathway, defined as chromosomal instability (CIN) responsible for up to 85% of CRC, and the serrated neoplasia pathway, accounting for the remaining 15%. CIN mechanisms involved chromosome alterations and DNA damage response network, affecting critical genes involved in cell function (APC, KRAS, PI3K, TP53) and pathways (WNT, MAPK, PI3K, TGF-β) [8]. The serrated neoplasia pathway is associated with RAS and BRAF gene mutations and epigenetic instability, characterized by the CpG island methylator phenotype (CIMP). Genome-wide studies identified new markers and phenotypic subtypes based on polymerase-mutations or mismatch repair deficiency leading to a hypermutated phenotype [8]. These two latter molecular events explain the microsatellite instability (MSI) identified in the 15–20% of CRCs [9].
Current biomarkers in mCRC treatment decision involve evidence of KRAS and NRAS mutations. A clear clinical meaning has only been achieved by KRAS oncogene in CRC patient management. KRAS oncogene regulates the activation of downstream effectors of several pathways, such as BRAF/RAS/MAPK, PI3K/AKT, RalGDS/p38-MAPK, etc., thus influencing normal cell physiology, neoplastic cell biology, and therapeutic responses. At least 40% of CRCs reported KRAS mutations that are biomarkers predictive of treatment efficacy and patient outcome [10]. Particularly, exon 2 KRAS mutations are correlated to advanced stage tumors and adverse prognosis [11]. Identification of KRAS mutations is a key molecular test for evaluating targeted therapies in mCRC. The presence of wild-type KRAS sequences guarantees the success of targeting by monoclonal antibodies (Cetuximab or Panitumumab) of the EGFR axis [12]. BRAF gene mutation (V600E) has been associated with aggressive clinical outcome in CRC patients [13]. Five percent of mCRC patients show a link between MSI-high (MSI-H) and a striking response to immune checkpoint blockade with anti-PD1 therapy [14]. Moreover, 3–8% of KRAS-wildtype CRC patients exhibit anti-EGFR therapy resistance explained by the presence of HER2 amplification.
Despite extensive knowledge of CRC biology and improvements in therapy, such as surgery, chemotherapy, and targeted therapies, this tumor remains one of the hard-to-treat cancers considering the high frequency of metastases and recurrences after surgery, and frequent resistance to first or second-line of treatment.
Cancer stem cells (CSCs) are multipotent and self-renewing cells whose role was first identified in hematologic malignancies and recently in solid tumors, including CRC [15]. Some evidence supports the hypothesis that onset, progression, and development of drug resistance in CRC might be related to the maintenance of a CSCs phenotype by deregulation of pathways involved in transformation, differentiation, growth, and epithelial to mesenchymal transitions (EMT) [16].
Recent data have validated the significant role of epigenetics in regulating the function of CRC cells [17] and colorectal cancer stem cells (CCSCs) [18]. Non-coding RNAs (ncRNAs) such as microRNAs (miRNAs) and long-non-coding RNAs (lncRNAs) control several cell functions and regulate gene expression by interacting with target mRNAs, resulting in either mRNA degradation or translational repression [19].
The relationship between miRNAs gene expression profile and CRC clinical outcome has been extensively analyzed [20]. MiRNAs control several cellular functions, including self-renewal and cellular differentiation, that make SCs insensitive to environmental stimuli that would normally stop most cells at the G1/S checkpoint. It could be proposed that the mechanisms used by SCs to overcome this checkpoint should be used by tumor cells to progress [21].
2. Cancer Stem Cells in Colorectal Cancer
Recent technological progresses have identified several genetic and epigenetic aberrations explaining the heterogeneous nature of CRC and justifying the requirement of personalized medicine for CRC patients. Additionally, CRC consists of heterogeneous cell populations differing for markers expression, proliferative capacity, morphology, and malignant potential. CRC intra-tumor heterogeneity is the seed of cancer formation due to the presence of CSCs. The context in which the tumor should be studied by applying the principles of stem cell biology is given by the clonal expansion and repopulation characteristics of the various cell lineages of CSCs within the tumor [22].
ISCs and transit-amplifying cells are the best candidates for the origin of CCSCs, given that their high proliferative capacity allows easy acquisition of genetic and epigenetic aberrations. Although, studies on histology of intestine and CRC suggested that CCCSs might arise from terminally differentiated cells [23]. Recent studies on de-differentiation process suggested that intestinal non-stem cell mutated and persevered could lead to adenoma progression in the presence of additional genomic events (e.g., b-catenin mutation combined with increased NFKB signaling or changes in the microenvironment) [24]. ISC features could be sustained by genetic/epigenetic aberrations acquired into different neoplastic subclones [25]. Stemness is responsible for three key processes that trigger tumor progression: Cell growth and proliferation, recurrence and metastasization, and therapy resistance (Figure 1).
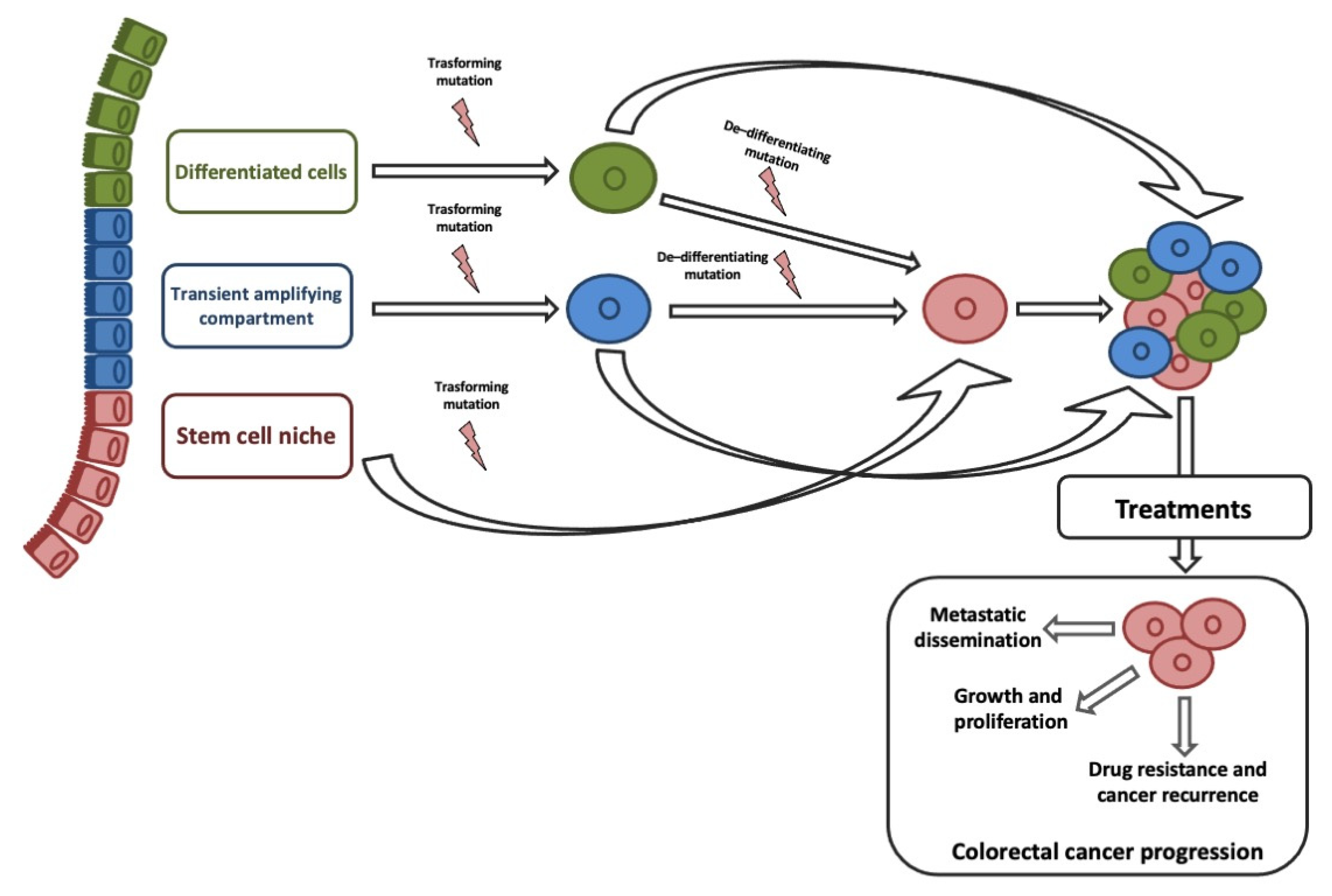
A convincing link between EMT and CSCs explained their crucial role in CRC progression and therapeutic resistance [26]. EMT-associated gene expression gives invasive and metastatic characteristics, resistance to therapies, and CSCs phenotypes on cancer cells [27][28][29][30]. The clinical implication is that by removing the CCSC, the tumor will no longer be capable of growing. Based on the CSC hypothesis, our diagnostic and therapeutic goals will be specifically discovered and targeted CCSCs [30].
Colorectal CSCs have been characterized based on specific markers. CD133 is well documented: It is a membrane-bound glycoprotein involved in primordial cell differentiation and EMT [31][32][33][34]. CD133+ cells produce IL-4 and use it to evade apoptosis. The treatment with IL-4-neutralizing antibody or with IL-4Rα antagonist will greatly enhance the sensitivity of CD133+ cells to chemotherapeutic drugs increasing antitumor efficacy, confirming previous observation [35]. CD133+ CRC cells manifested the CSC-like properties, such as higher levels of SC markers OCT4 and SOX2, tumor sphere forming ability, and more tumorigenic in NOD/SCID mice [36], which is consistent with OCT4 and SOX2 overexpression in poorly differentiated human tumors [37]. CSCs display EMT phenotype, possess high levels of transcription factors SNAIL and TWIST, mesenchymal markers vimentin and fibronectin, and low levels of epithelial protein E-cadherin. CD133+ CSCs might escape immune surveillance by expressing inhibitory molecule B7H1. The EMT phenotype was determined in human CRC cells in which CD133 was co-expressed with B7H1 [36]. All these findings do speculate that CD133+ CCSCs showing EMT phenotype and co-expressing B7H1 may evade immune surveillance. Schmohl et al. demonstrated that innate immune system can be effectively recruited to kill CCSCs using bispecific antibodies targeting CD133. An innovative engineering technology has developed a new anti-cancer molecule. Two fully humanized single chain DNA fragment variable antibodies, recognizing CD16 on NK-cells and CD133 on CSC, were spliced creating a novel drug defined 16 × 133 novel bispecific killer cell engager (BiKE). This molecule simultaneously recognizes antigens to facilitate an immunologic synapse. 16×133 BiKE is a potent engager of the innate immune system capable of inducing NK-cell degranulation and IFN-γ production and mediating selective targeting of CD133+ CSCs. 16×133 BiKE may have therapeutic potential in a clinical NK-cell therapy program for carcinomas, as it could serve as an alternative therapy for drug resistant CSCs by its unique mechanism of action [38].
Analysis of CCSC lysate indicates that they may be a sufficient resource of tumor antigens, and CCSC lysate-based vaccines could stimulate proliferation and differentiation of immune cells overcoming the weak immunogenicity of CCSCs [39][40]. Mice subcutaneously immunized with the CCSC lysate reduced the tumor growth via a target killing of CCSCs. They decrease CD133+ and ALDH+ cells in tumors vs. control vaccine groups, resulting in elevated NK cytotoxicity, perforin production, granzyme B, IFN-γ, memory B cells, and anti-MUC1 antibodies. MUC1 could induce metastasis, cell invasion and proliferation, drug resistance, and angiogenesis in CRC [41]. Further, the antitumor efficacy of CCSC vaccine in MUC1 knockdown was partially impaired [42].
ALDH1+ cells are found at low levels at crypt bases, but increase during progression from normal to APC-mutant adenoma [43]. A high percentage of tumor cells expressing ALDH1 correlate with poor prognosis in various cancers, and display properties of CSCs. ALDH1 is a promising marker to identify CCSCs and a potential candidate for CSC-directed therapy, due to the low expression of ALDH1 in normal colon compared to tumor [44][45].
CD44 is a cell surface glycoprotein involved in cell–cell interactions, adhesion of the cytoskeleton to the extracellular matrix and cell migration [15][46] and, depending on WNT signaling, its overexpression is an early event in the transformation of adenoma to CRC. CD44+/CD24−/CD133− cells form the most aggressive colon tumors, removing the requirement of CD133 in tumor onset [47]. Numerous studies have isolated and characterized EpCAM+/CD44+ cells from CRCs. Dalerba et al. found that normal colon and CRC both contain two populations of cells: EpCAMHigh/CD44+ and EpCAMLow/CD44−. Only the first one develops tumors when injected into non-obese diabetic/SCID mice [15]. Using a FACS-based selection, Kemper et al. identified in primary CRCs, a small population of EpCAM+/LGR5+ cells. Spheroid cultures resulting from primary CRC are enriched for CSCs and express high levels of LGR5, while cellular differentiation reduces LGR5 expression. The LGR5high CRC cells are more clonogenic and tumorigenic than LGR5low CRC cells. LGR5 overexpression results in higher clonogenic growth, indicating that LGR5 is a new functional marker for CCSCs [48].
OCT4, SOX2, and NANOG are transcription factors that play a main role in the regulation of pluripotency and implement the self-regulation of their expressions by linking to their promoting regions [49]. OCT4 influences embryogenesis, stem cell maintenance, tumor growth, and metastasis [49][50]. Though it is an important CSC marker, its expression has also been noted in colonic normal tissue. OCT4 expression has been principally identified in the cytoplasm of CRC cells, suggesting a drive factor of recurrence, presumably by preventing apoptosis [50]. OCT4 expression in CRC was present in cells which are undergoing EMT, a key stage in progression and metastasis, and increasing the cell stem-like phenotype [51]. SOX2 is reported to prevent differentiation of neural progenitor cells and to be overexpressed in CRC stage III [50]. Talebi et al. comparing normal colon, dysplastic polyps, and adenocarcinomas identified a significant correlation between SOX2 expression and CRC [52]. High level of SOX2 in CRCs positively correlates with metastases and lymph node infiltration. SOX2 controls OCT4 expression, and this combination of transcription factors promote pluripotency [49]. NANOG also influences pluripotency through transcriptional control [50]. Ibrahim et al. found NANOG in a subpopulation of colon epithelial cells in primary CRC. The OCT4/SOX2 complex regulates NANOG expression and controls the expression of pluripotency-related genes [53]. The high expression of NANOG is correlated with poor prognosis in CRC patients [49].
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA. Cancer J. Clin. 2018, 68, 394–424.
- Arnold, M.; Sierra, M.S.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global patterns and trends in colorectal cancer incidence and mortality. Gut 2017, 66, 683–691.
- Perrott, S.; Laurie, K.; Laws, K.; Johnes, A.; Miedzybrodzka, Z.; Samuel, L. Young-onset colorectal cancer in the North East of Scotland: Survival, clinico-pathological features and genetics. BMC Cancer 2020, 20, 108.
- Pira, G.; Uva, P.; Scanu, A.M.; Rocca, P.C.; Murgia, L.; Uleri, E.; Piu, C.; Porcu, A.; Carru, C.; Manca, A.; et al. Landscape of transcriptome variations uncovering known and novel driver events in colorectal carcinoma. Sci. Rep. 2020, 10, 432.
- Uleri, E.; Piu, C.; Caocci, M.; Ibba, G.; Sanges, F.; Pira, G.; Murgia, L.; Barmina, M.; Giannecchini, S.; Porcu, A.; et al. Multiple Signatures of the JC Polyomavirus in Paired Normal and Altered Colorectal Mucosa Indicate a Link with Human Colorectal Cancer, but Not with Cancer Progression. Int. J. Mol. Sci. 2019, 20, 5965.
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502.
- Riihimaki, M.; Hemminki, A.; Sundquist, J.; Hemminki, K. Patterns of metastasis in colon and rectal cancer. Sci. Rep. 2016, 6.
- Muzny, D.M.; Bainbridge, M.N.; Chang, K.; Dinh, H.H.; Drummond, J.A.; Fowler, G.; Kovar, C.L.; Lewis, L.R.; Morgan, M.B.; Newsham, I.F.; et al. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337.
- Marzouk, O.; Schofield, J. Review of histopathological and molecular prognostic features in colorectal cancer. Cancers 2011, 3, 2767–2810.
- Bahrami, A.; Hassanian, S.M.; ShahidSales, S.; Farjami, Z.; Hasanzadeh, M.; Anvari, K.; Aledavood, A.; Maftouh, M.; Ferns, G.A.; Khazaei, M.; et al. Targeting RAS signaling pathway as a potential therapeutic target in the treatment of colorectal cancer. J. Cell. Physiol. 2018, 233, 2058–2066.
- Li, W.; Qiu, T.; Zhi, W.; Shi, S.; Zou, S.; Ling, Y.; Shan, L.; Ying, J.; Lu, N. Colorectal carcinomas with KRAS codon 12 mutation are associated with more advanced tumor stages. BMC Cancer 2015, 15.
- Amado, R.G.; Wolf, M.; Peeters, M.; Van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol. 2008, 26, 1626–1634.
- Van Cutsem, E.; Köhne, C.H.; Láng, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol. 2011, 29, 2011–2019.
- Le, D.T.; Uram, J.N.; Wang, H.; Bartlett, B.R.; Kemberling, H.; Eyring, A.D.; Skora, A.D.; Luber, B.S.; Azad, N.S.; Laheru, D.; et al. PD-1 Blockade in Tumors with Mismatch-Repair Deficiency. N. Engl. J. Med. 2015, 372, 2509–2520.
- Dalerba, P.; Dylla, S.J.; Park, I.K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic characterization of human colorectal cancer stem cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163.
- Frank, N.Y.; Schatton, T.; Frank, M.H. The therapeutic promise of the cancer stem cell concept. J. Clin. Investig. 2010, 120, 41–50.
- Fadda, A.; Gentilini, D.; Moi, L.; Barault, L.; Leoni, V.P.; Sulas, P.; Zorcolo, L.; Restivo, A.; Cabras, F.; Fortunato, F.; et al. Colorectal cancer early methylation alterations affect the crosstalk between cell and surrounding environment, tracing a biomarker signature specific for this tumor. Int. J. Cancer 2018, 143, 907–920.
- Li, F.; Xu, Y.; Xu, X.; Ge, S.; Zhang, F.; Zhang, H.; Fan, X. lncRNA HotairM1 Depletion Promotes Self-Renewal of Cancer Stem Cells through HOXA1-Nanog Regulation Loop. Mol. Ther. Nucleic Acids 2020, 22, 456–470.
- Guo, L.; Lu, Z. The Fate of miRNA* Strand through Evolutionary Analysis: Implication for Degradation As Merely Carrier Strand or Potential Regulatory Molecule? PLoS ONE 2010, 5, e11387.
- Baek, D.W.; Kim, G.; Kang, B.W.; Kim, H.J.; Park, S.Y.; Park, J.S.; Choi, G.S.; Kang, M.K.; Hur, K.; Kim, J.G. High expression of microRNA-199a-5p is associated with superior clinical outcomes in patients with locally advanced rectal cancer. J. Cancer Res. Clin. Oncol. 2020, 146, 105–115.
- Croce, C.M.; Calin, G.A. miRNAs, cancer, and stem cell division. Cell 2005, 122, 6–7.
- Shimokawa, M.; Ohta, Y.; Nishikori, S.; Matano, M.; Takano, A.; Fujii, M.; Date, S.; Sugimoto, S.; Kanai, T.; Sato, T. Visualization and targeting of LGR5 + human colon cancer stem cells. Nature 2017, 545, 187–192.
- Shih, I.M.; Wang, T.L.; Traverso, G.; Romans, K.; Hamilton, S.R.; Ben-Sasson, S.; Kinzler, K.W.; Vogelstein, B. Top-down morphogenesis of colorectal tumors. Proc. Natl. Acad. Sci. USA 2001, 98, 2640–2645.
- Schwitalla, S.; Fingerle, A.A.; Cammareri, P.; Nebelsiek, T.; Göktuna, S.I.; Ziegler, P.K.; Canli, O.; Heijmans, J.; Huels, D.J.; Moreaux, G.; et al. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell 2013, 152, 25–38.
- Iyer, D.N.; Sin, W.Y.; Ng, L. Linking stemness with colorectal cancer initiation, progression, and therapy. World J. Stem Cells 2019, 11, 519–534.
- Fan, F.; Samuel, S.; Evans, K.W.; Lu, J.; Xia, L.; Zhou, Y.; Sceusi, E.; Tozzi, F.; Ye, X.C.; Mani, S.A.; et al. Overexpression of Snail induces epithelial-mesenchymal transition and a cancer stem cell-like phenotype in human colorectal cancer cells. Cancer Med. 2012, 1, 5–16.
- Dalerba, P.; Clarke, M.F. Cancer Stem Cells and Tumor Metastasis: First Steps into Uncharted Territory. Cell Stem Cell 2007, 1, 241–242.
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008, 133, 704–715.
- Hollier, B.G.; Evans, K.; Mani, S.A. The epithelial-to-mesenchymal transition and cancer stem cells: A coalition against cancer therapies. J. Mammary Gland Biol. Neoplasia 2009, 14, 29–43.
- Pardal, R.; Clarke, M.F.; Morrison, S.J. Applying the principles of stem-cell biology to cancer. Nat. Rev. Cancer 2003, 3, 895–902.
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. Expression of CD133 enriches for colon cancer stem cells. Ann. Surg. Oncol. 2007, 14, 22.
- Ieta, K.; Tanaka, F.; Haraguchi, N.; Kita, Y.; Sakashita, H.; Mimori, K.; Matsumoto, T.; Inoue, H.; Kuwano, H.; Mori, M. Biological and genetic characteristics of tumor-initiating cells in colon cancer. Ann. Surg. Oncol. 2008, 15, 638–648.
- Vaiopoulos, A.G.; Kostakis, I.D.; Koutsilieris, M.; Papavassiliou, A.G. Concise review: Colorectal cancer stem cells. Stem Cells 2012, 30, 363–371.
- Todaro, M.; Francipane, M.G.; Medema, J.P.; Stassi, G. Colon Cancer Stem Cells: Promise of Targeted Therapy. Gastroenterology 2010, 138, 2151–2162.
- Todaro, M.; Alea, M.P.; Di Stefano, A.B.; Cammareri, P.; Vermeulen, L.; Iovino, F.; Tripodo, C.; Russo, A.; Gulotta, G.; Medema, J.P.; et al. Colon Cancer Stem Cells Dictate Tumor Growth and Resist Cell Death by Production of Interleukin-4. Cell Stem Cell 2007, 1, 389–402.
- Zhi, Y.; Mou, Z.; Chen, J.; He, Y.; Dong, H.; Fu, X.; Wu, Y. B7H1 Expression and Epithelial-To-Mesenchymal Transition Phenotypes on Colorectal Cancer Stem-Like Cells. PLoS ONE 2015, 10, e0135528.
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507.
- Schmohl, J.U.; Gleason, M.K.; Dougherty, P.R.; Miller, J.S.; Vallera, D.A. Heterodimeric Bispecific Single Chain Variable Fragments (scFv) Killer Engagers (BiKEs) Enhance NK-cell Activity Against CD133+ Colorectal Cancer Cells. Target. Oncol. 2016, 11, 353–361.
- Pan, T.; Xu, J.; Zhu, Y. Self-renewal molecular mechanisms of colorectal cancer stem cells. Int. J. Mol. Med. 2017, 39, 9–20.
- Zeuner, A.; Todaro, M.; Stassi, G.; De Maria, R. Colorectal cancer stem cells: From the crypt to the clinic. Cell Stem Cell 2014, 15, 692–705.
- Rajabi, H.; Hiraki, M.; Kufe, D. MUC1-C activates polycomb repressive complexes and downregulates tumor suppressor genes in human cancer cells. Oncogene 2018, 37, 2079–2088.
- Guo, M.; Luo, B.; Pan, M.; Li, M.; Zhao, F.; Dou, J. MUC1 plays an essential role in tumor immunity of colorectal cancer stem cell vaccine. Int. Immunopharmacol. 2020.
- Huang, E.H.; Hynes, M.J.; Zhang, T.; Ginestier, C.; Dontu, G.; Appelman, H.; Fields, J.Z.; Wicha, M.S.; Boman, B.M. Aldehyde dehydrogenase 1 is a marker for normal and malignant human colonic stem cells (SC) and tracks SC overpopulation during colon tumorigenesis. Cancer Res. 2009, 69, 3382–3389.
- Deng, S.; Yang, X.; Lassus, H.; Liang, S.; Kaur, S.; Ye, Q.; Li, C.; Wang, L.P.; Roby, K.F.; Orsulic, S.; et al. Distinct expression levels and patterns of stem cell marker, aldehyde dehydrogenase isoform 1 (ALDH1), in human epithelial cancers. PLoS ONE 2010, 5.
- Tomita, H.; Tanaka, K.; Tanaka, T.; Hara, A. Aldehyde dehydrogenase 1A1 in stem cells and cancer. Oncotarget 2016, 7, 11018–11032.
- Morath, I.; Hartmann, T.N.; Orian-Rousseau, V. CD44: More than a mere stem cell marker. Int. J. Biochem. Cell Biol. 2016, 81, 166–173.
- Shmelkov, S.V.; Butler, J.M.; Hooper, A.T.; Hormigo, A.; Kushner, J.; Milde, T.; St Clair, R.; Baljevic, M.; White, I.; Jin, D.K.; et al. CD133 expression is not restricted to stem cells, and both CD133 + and CD133- metastatic colon cancer cells initiate tumors. J. Clin. Investig. 2008, 118, 2111–2120.
- Kemper, K.; Prasetyanti, P.R.; De Lau, W.; Rodermond, H.; Clevers, H.; Medema, J.P. Monoclonal antibodies against Lgr5 identify human colorectal cancer stem cells. Stem Cells 2012, 30, 2378–2386.
- Hadjimichael, C.; Chanoumidou, K.; Papadopoulou, N.; Arampatzi, P.; Papamatheakis, J.; Kretsovali, A. Common stemness regulators of embryonic and cancer stem cells. World J. Stem Cells 2015, 7, 1150–11584.
- Amini, S.; Fathi, F.; Mobalegi, J.; Sofimajidpour, H.; Ghadimi, T. The expressions of stem cell markers: Oct4, Nanog, Sox2, nucleostemin, Bmi, Zfx, Tcl1, Tbx3, Dppa4, and Esrrb in bladder, colon, and prostate cancer, and certain cancer cell lines. Anat. Cell Biol. 2014, 47, 1–11.
- Dai, X.; Ge, J.; Wang, X.; Qian, X.; Li, X. OCT4 regulates epithelial-mesenchymal transition and its knockdown inhibits colorectal cancer cell migration and invasion. Oncol. Rep. 2013, 29, 155–160.
- Talebi, A.; Kianersi, K.; Beiraghdar, M. Comparison of gene expression of SOX2 and OCT4 in normal tissue, polyps, and colon adenocarcinoma using immunohistochemical staining. Adv. Biomed. Res. 2015, 4, 234.
- Ibrahim, E.E.; Babaei-Jadidi, R.; Saadeddin, A.; Spencer-Dene, B.; Hossaini, S.; Abuzinadah, M.; Li, N.; Fadhil, W.; Ilyas, M.; Bonnet, D.; et al. Embryonic NANOG activity defines colorectal cancer stem cells and modulates through AP1- and TCF-dependent mechanisms. Stem Cells 2012, 30, 2076–2087.


