 +1 credit
+1 credit
| Version | Summary | Created by | Modification | Content Size | Created at | Operation |
|---|---|---|---|---|---|---|
| 1 | Mark Miles | + 2573 word(s) | 2573 | 2021-06-23 05:35:44 | | | |
| 2 | Dean Liu | Meta information modification | 2573 | 2021-07-12 05:00:04 | | | | |
| 3 | Dean Liu | Meta information modification | 2573 | 2021-07-12 05:00:42 | | |
Video Upload Options
An important role of cell death pathways is to protect tissues and minimize disease by limiting the transference of potentially oncogenic mutations to daughter clones. However, there is increasing evidence demonstrating that activation of sublethal cell death signaling pathways, in particular apoptotic signaling, in the absence of direct DNA damaging stimuli, can promote genomic instability in cells that fail to die. This may increase the risk of the formation of subsequent neoplasms. Apoptosis-mediated mutagenesis occurs indirectly via sublethal activation of caspases and apoptotic nucleases (specifically CAD). On the other hand, cells surviving sublethal necroptotic signaling did not acquire mutations, most likely due to caspase-independent pathways, although the possibility of mutagenesis under conditions of oxidative stress are still elusive. It may therefore be possible for necroptosis-inducing anti-cancer drugs to be less likely than apoptosis-inducing or DNA damaging drugs to trigger therapy-related cancers.
1. Introduction
Over the years, much research has been invested into understanding and defining the molecular mechanisms of various cell death pathways. A better understanding will enable us to develop effective therapeutics to combat diseases that are underscored by dysregulated cell death. A variety of mechanisms exist by which a cell can regulate its demise. Redundant cellular self-destruction pathways may act as failsafe switches for cells in contexts where blocks prevent sufficient propagation of one or more cell death pathways. Perturbations in cells’ responsiveness to cell death signaling can lead to disease states, many of which are driven by mutations that fuel this dysregulation. Mutations arise upon incorrect repair of damaged DNA. The genomic instability brought on by various mutations may prime cells to a “mutation-prone” state, further allowing the acquisition of more mutations and facilitating oncogenic transformation. In this aspect, cell death pathways act to protect tissues by limiting the transference of potentially oncogenic mutations to daughter clones. However, there is increasing evidence demonstrating that activation of sublethal cell death signaling pathways, in particular apoptotic signaling, in the absence of direct DNA-damaging stimuli, can promote genomic instability in cells that fail to die. We will look at DNA damage from a different viewpoint: extensive DNA damage initiated by radiation or chemotherapeutics initiates cell death, but nucleases activated during apoptosis (or some other forms of cell death) can also inflict genomic damage that may be mis-repaired.
2. Mutagenic Consequences of Apoptotic Signaling
Our knowledge of apoptotic cell death is extensive. Apoptosis is known for its “silent” mode of cell destruction without triggering immune activation. It is crucial during embryonic development, for maintaining tissue homeostasis under threatening conditions, and for the removal of cells that have reached their lifespan. The pathological impact of apoptosis can have various effects on tumorigenesis and cancer. In the first instance, caspase activity within cells that do succumb to apoptotic death can offer oncogenic advantages. Dying cells were reported to release mitogenic signals, via active executioner caspases, which promoted the proliferation of surviving, neighboring tumor cells. This phenomenon termed “apoptosis-induced proliferation” has implications in tumor repopulation, post-therapeutic relapse and oncogenic cellular evolution. In the second instance, sublethal apoptotic signaling could provoke genomic instability via mutagenesis (Figure 1), potentially leading to the oncogenic transformation of non-cancerous cells and increasing the risk of de novo or subsequent cancer formation.
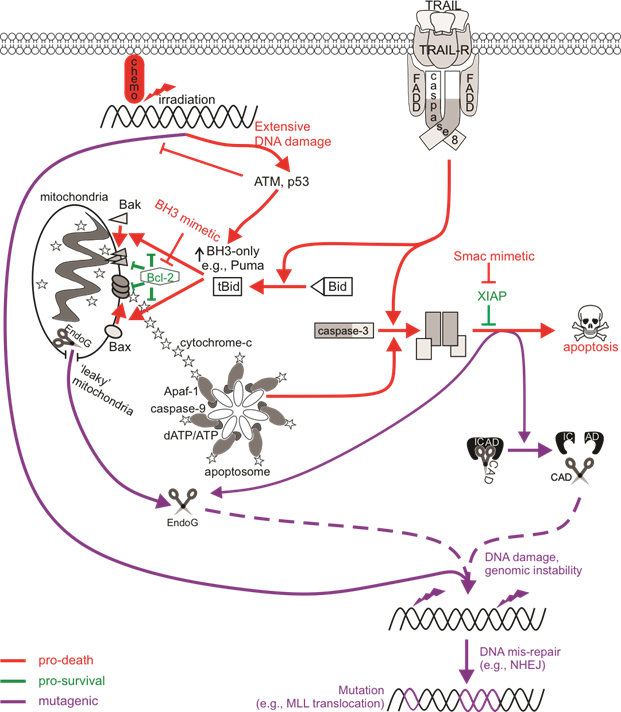
Figure 1. The mutagenic potential of apoptotic signaling. Chemotherapy drugs induce DNA damage that can be recognized by DNA damage sensors such as ATM to direct cell signaling towards DNA repair or death. An insufficient response (for instance, when the ATM function is defective) may encourage the activation of low-fidelity repair pathways such as non-homologous end joining (NHEJ) that are error-prone, increasing the likelihood of genomic mutations. Cell death may also be primed for, leading to p53-mediated upregulation of pro-apoptotic Bcl-2 proteins to induce Bax/Bak-mediated permeabilization of the mitochondrial outer membrane (MOMP), cytochrome c release, and the activation of caspase-9 via the apoptosome. Active caspase-9 can promote executioner caspase activation, which then cleave cellular substrates to induce apoptosis. Extrinsic activation of death receptors can also activate this caspase-cascade. Apoptosis-induced genomic fragmentation by caspase-activated DNase (CAD), which becomes active upon executioner-mediated cleavage of its inhibitor ICAD, can provoke mutations via NHEJ repair if sublethal levels of apoptosis are achieved. BH3 mimetics promote apoptotic cell death by relieving the inhibition of pro-survival Bcl-2 proteins. Some cells may experience “minority” MOMP, which describes the sublethal release of cytochrome c and caspase activation from “leaky” mitochondria. Mutagenesis upon “minority” MOMP can occur due to the release of endonuclease G (EndoG) from the mitochondria and its direct action on DNA or via caspase/CAD-dependent pathways. The mis-repair of nuclease-mediated DNA fragmentation could lead to oncogenic mutations such as chromosomal rearrangements that alter the MLL gene to increase the risk of acute myeloid leukemia.
3. Possible Mutagenic Consequences of Necroptotic Signaling
Necroptotic cell death is a form of regulated necrotic death that stimulates immune activation. Depending on the status or expression of certain downstream proteins, TNFα-mediated activation of its receptors can stimulate pro-survival signaling pathways, apoptosis, or necroptosis, and these are largely mediated by receptor-interacting protein (RIP) kinases. In the absence of pro-survival signaling (such as when cIAP1/2 is degraded by Smac/DIABLO or drugs that mimic its function), RIPK1 is deubiquitinated and instead associates with FADD and caspase-8, transitioning from pro-survival complex I to pro-apoptotic complex IIa. Given that necroptosis is a caspase-independent form of cell death, it may represent an effective anti-tumor alternative to classical anti-cancer therapies that activate apoptotic machineries that may be defective in chemo-resistant cells, and may also avoid the mutagenic and possibly oncogenic effects of sublethal caspase signaling discussed earlier.
Much research is currently being carried out to fully understand all aspects of necroptosis, so relatively little is presently known about the direct mutagenic consequences of necroptotic signaling (Figure 2). MLKL-mediated membrane rupture was initially considered a “point of no return” in cell necroptotic fate. However, just like the previous notion that caspase activation was considered a lethal event (which we now accept is not the case as cells can withstand sublethal levels of active caspases), emerging evidence challenges this ‘always-fatal’ function of MLKL. In this context, the ESCRT machinery most likely engages membrane repair as ESCRT components localized at MLKL damage sites on the plasma membrane, and silencing of ESCRT genes sensitized cells to necroptosis. This delayed or even prevented MLKL-mediated loss of plasma membrane integrity, enabling cell survival following necroptosis. Another way in which a cell could conceivably withstand active MLKL may be through its sequestration and release in necroptotic bodies. A proposed physiological role for delayed necroptotic death may be to maximize immune stimulation by allowing extra time for immunogenic signals to attract immune cells, for example with longer PS exposure or the release of pro-inflammatory cytokines, prior to complete demise of the cell. Given the evidence describing cell “resuscitation” following necroptosis, are there mutagenic consequences to sublethal necroptotic signaling? Recent in vitro analysis determined that classic activation of necroptosis via TNFα, caspase-8 inhibition, and IAP antagonism failed to provoke DNA damage or mutations in surviving cells. Importantly, DNA damage was not detected in cells expressing a lethal constitutively active MLKL mutant, implying that the execution of necroptosis by MLKL was not genotoxic.
While survival from non-genotoxic necroptotic stimuli may be a promising non-mutagenic feature of necroptosis, the activation of upstream necroptotic components upon necroptotic signaling may have unwanted consequences that may contribute to cancer initiation and progression or other disease. Redox regulatory roles of necroptosis have been reported, and thus the oxidative stress associated with these processes may have mutagenic consequences. While this may be an important physiological response in pathogen-infected cells, for instance by promoting inflammasome activation, ROS modulation by necroptosis may contribute pathologies related to oxidative stress . In addition, high levels of ROS can damage DNA via direct reactivity to the sugar backbone of DNA, thereby oxidizing nucleoside bases or modulating replication stress.
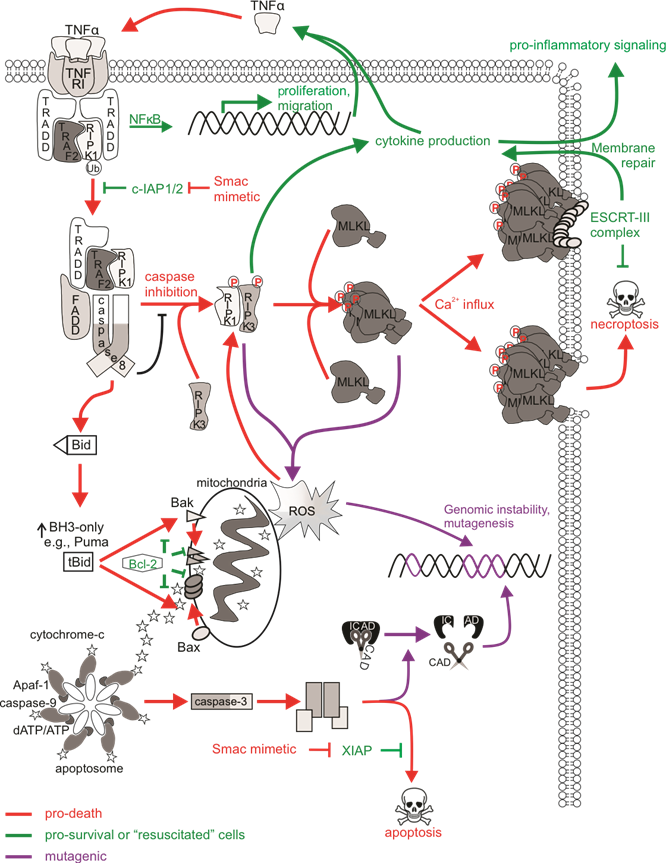
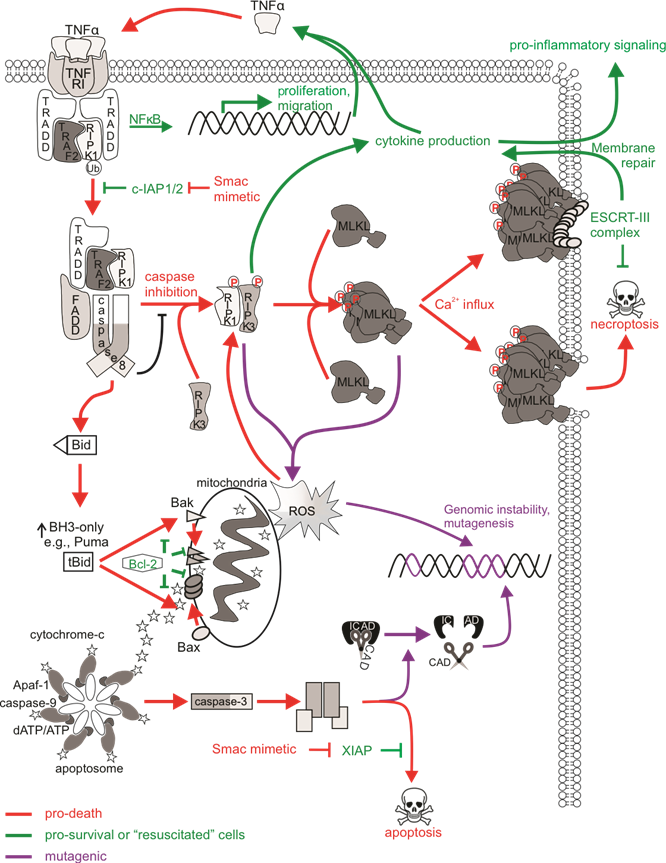
Figure 2. The mutagenic potential of necroptotic signaling. cIAP1/2 acting at TNFR1 complex I polyubiquitinates RIPK1 to allow for NFκB transcription and expression of proliferative and migratory genes. Some cells can autocrine produce TNFα to further stimulate TNFR1 signaling. There is a transition to pro-death complexes IIa (if caspase-8 is present) or IIb (if caspase-8 activity is inhibited) upon incapacitation of cIAP1/2. Pro-apoptotic ripoptosome formation leads to MOMP and caspase activation, which can provoke mutations via CAD-dependent DNA damage. The pro-necroptotic necrosome is formed in cells lacking caspase-8 activity allowing RIPK1 recruitment of RIPK3, the autophosphorylation of RIPK3, and the subsequent phosphorylation and activation of MLKL by RIPK3. Activated MLKL oligomerizes then translocates to the plasma membrane, where it forms pores to compromise membrane integrity. The ensuing MLKL-mediated necroptotic death is not associated with DNA damage. The initiation of ESCRT-III-mediated membrane repair of MLKL pores delays the onset of or even prevents necroptotic cell lysis and allows for RIPK3-dependent cytokine production and release. Survival following necroptotic signaling is known as “resuscitation”. The necrosome can also stimulate mitochondrial-dependent or independent ROS production, which can stabilize the RIPK1/RIPK3 necrosome as a positive feedback loop. DNA can be directly impacted by ROS, and mutagenesis might occur if damage is mis-repaired or there is sustained genomic instability in “resuscitated” cells.
3. Possible Mutagenic Consequences of Sublethal Pyroptotis Signaling
Pyroptosis is a pro-inflammatory mode of cell death which acts as a defense mechanism against infection. It is the culmination of molecular pathways that respond to the activation of nucleotide-binding oligomerization (NOD)- like receptors (NLRs), which are pattern recognition receptors (PRRs) that sense internal danger signals, such as pathogen-associated molecular patterns (PAMPs) and DAMPs. TLRs, C-type lectins, and galectins fall under the NLR family of PRRs and participate in molecular complexes termed “inflammasomes” to stimulate immune activity and pyroptotic cell death. Active caspase-1 cleaves pro-IL-1β into its mature form as well as cytosolic gasdermin D (GSDMD), enabling the oligomerization of its N-terminal fragment, which forms ring-like pores on the plasma membrane to allow cytokine release and facilitate pyroptotic cell lysis. Caspase-8 also contributes to canonical and non-canonical NLRP3 inflammasomes in the absence of caspase-1.
GSDMD-generated pores were initially believed to be sufficient for pyroptosis and concurrent IL-1β release, however the release of IL-1 cytokines from inflammasome-active viable cells, also known as “hyperactivated” cells, can also occur in the absence of pyroptosis, suggesting that mechanisms exist to regulate cell fate after inflammasome activation. This scenario—viable cells bearing active inflammasomes—could be considered “sublethal pyroptotic signaling”. This highlights the ability of cells to withstand inflammasome activation and inflammatory caspase activity, and some downstream processes, without succumbing to cell death.
The fact that GSDMD membrane pores can form in viable cells to allow cytokine release suggests that a threshold amount of activated GSDMD needs to be achieved in order to execute pyroptosis. Similar to the restoration of plasma membrane integrity in MLKL activated “resuscitated” necroptotic cells discussed earlier, ESCRT-mediated membrane repair can also follow GSDMD activation and pore formation, resulting in the delay or blockage of pyroptosis, and enhancing cell survival. DNA damage has been detected in cells bearing inflammasome activity. However, whether this leads to mutations in surviving cells, or merely reflects DNA degradation in dying cells, has not been determined. The mechanism responsible for the DNA damage is also unclear. Intact ICAD was detected in treated and untreated cells, prompting researchers to argue that CAD was not activated via the inflammasome. Apoptotic caspases can also be activated upon inflammasome formation. Caspase-8 proteolytic activation and catalytic activity was detected upstream of NLRP3 activation, suggesting that caspase-3 processing by caspase-8 could occur, although caspase-3 mediated apoptotic signaling was not detected. These suggest that the inflammasomes can activate DNA repair mechanisms to repair ROS-generated or possibly even CAD- or EndoG-mediated DNA damage in a caspase-3-dependent or independent manner. It is therefore possible for caspase-1-mediated sublethal activation of caspase-3 to promote genomic instability in inflammasome active, non-pyroptotic cells if NLRP3-mediated activation of DNA repair pathways initiates mis-repair in error-prone cells (such as those with defective high-fidelity repair) surviving pyroptotic signaling (Figure 3).
4. Concluding Remarks
Improvements in outcomes for some cancer types have prompted clinicians and researchers to focus on the more nuanced goal of sparing cured patients severe acute and late adverse effects of therapy. A well-recognized late effect of anti-cancer treatment is subsequent cancers, which can arise, at least in part, due to the mutational activity of traditional anti-cancer therapies. Ionizing radiation and certain chemotherapeutic drug classes, such as topoisomerase-II poisons, induce chromosomal changes that enhance a patient’s risk of subsequent oncogenesis. It is now possible to trigger cancer cell death directly, by engaging cell death pathways, rather than indirectly by creating DNA damage which secondarily stimulates cancerous cells to die. This prospect raised the possibility that therapy-induced cancers could be avoided. Unfortunately, this hope has been tampered, at least as far as direct apoptosis-inducing drugs are concerned, by the realization that pro-apoptotic stimuli can promote genomic instability via caspase-mediated activation of nucleases (Figure 4).
Drugs that promote death by necroptosis may avoid many of the pro-survival advantages that apoptosis-resistant cancer cells rely on. Due to this, these drugs may also be less likely to exert the same selective pressure as chemotherapy drugs, which were described to boost the frequency of pre-existing clones harboring pro-cancerous mutations in tumor suppressor genes such as TP53. The observation that sublethal necroptotic signaling was non-mutagenic provides hope that drugs which destroy cancer cells via necroptosis may be less prone to trigger development of second cancers than chemotherapy or radiotherapy, or possibly even than direct apoptosis inducers. This feature may be especially beneficial for cancer patients with germline flaws in DNA damage response pathways, for whom mutagenic therapies would be particularly risky. As yet, no anti-cancer agents have been created that exclusively trigger necroptosis, but Smac mimetic treatment (which can provoke apoptotic or necroptotic cell death) was non-mutagenic in vitro, even when accurate DNA repair was compromised. Further research will be needed to define the mutagenicity associated with sublethal activation of more recently described cell death pathways, including pyroptosis and ferroptosis, and the prospects for therapeutic exploitation of these pathways.
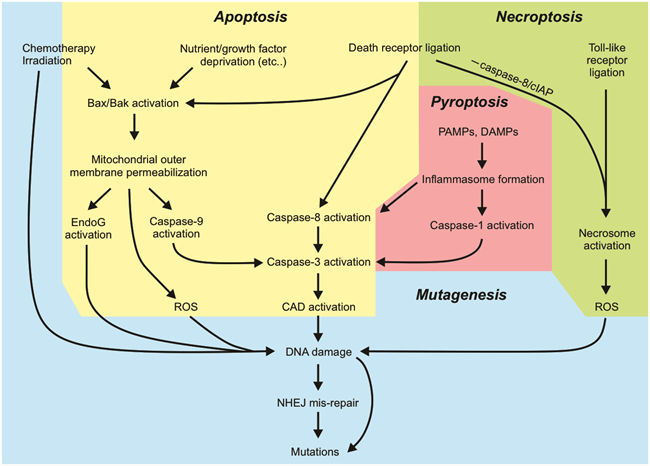
Figure 4. Summary of the key aspects of cell death pathways that can lead to mutagenesis.


