1. Bioinformatics Predictions of Ion Channel Structures and Functions
X-ray crystallography, NMR data, etc. on transmembrane proteins are generally used to predict the optimal protein structures. These techniques require the use of extremely expensive necessary ingredients and a tuned laboratory setup. Bioinformatics modeling utilizing appropriate techniques that may promote in silico mechanics and energetics of the protein structure considering the underlying mechanisms are often popularly considered in biophysical studies of proteins. Membrane proteins are generally studied specifically to address their ion channel-forming potency. Bioinformatics techniques play crucial roles when important molecular actions are to be inspected to explain the experimental facts obtained in vitro studies, such as their imaging in the interface of hydrophobic/hydrophilic regions, electrophysiology record of currents across membranes hosting the proteins, etc. Molecular dynamics (MD) simulations often appear as important computational techniques to detect energetics underlying biomolecular interactions. We have been quite successful in biophysical addressing, using MD simulations, of the channel energetics involving channel subunits and membrane lipids for small channels, such as gramicidin A, alamethicin, and chemotherapy drug-induced channels in model membrane systems
[1][2][3][4][5][6]. In these publications altogether we could establish a single fundamental fact that the channel stability inside the membrane is due to nothing but molecular mechanisms depending on charge-based screened Coulomb interaction energetics among functional charge groups in the ion channel complex involving channel subunit peptides or drugs and membrane lipids. Our computational in silico assays (numerical computations and MD simulations) simply supported the experimental findings in the distance and time-dependent channel subunit-lipid interaction energetics theoretically. We could calculate the binding energies and evaluate the binding energetics in the channel complex and thus know of the statistical mechanical nature in the channel stability in a biological thermodynamic environment. The readers are invited to read directly from these articles to gain further insights.
Besides various computational assays addressing the general structure and function of channel proteins, bioinformatics templates that draw information from various databank on the channel protein structures, genomics of the proteins, mutations in genes of the ion channel proteins are found to produce crucial information about channel functions in both healthy cells and mutated (disease) conditions.
The aspects addressing the ion channel protein genetics and mutations are presented later in this article using a few example case studies. Here we wish to address the general aspects of ion channel structures and functions using bioinformatics techniques
[7] including various computational assays and in silico modeling.
Table 1 presents a set of ion channels that are addressed using various in silico computational techniques
[8].
Table 1. Ion Channel modeling and simulation studies. The references quoted in the table are readily found as referenced in article
[8]. Here the general area of ion channels are organized according to the system type and computational models employed. “Reprinted (adapted) with permission from
[8]”.
| System |
Method
(We Quote Here the References, Numbered in ref. [8]. We Avoided to Relist the Huge Amount of References Here.) |
| Continuum |
Implicit Solvent MD |
All-Atom MD |
Hybrid |
CG |
Others (QM) |
| gramcidins |
8–15 |
16, 17 |
18–31 |
52, 53 |
|
54 |
| Other membrane porins |
55–61 |
55, 62–64 |
55, 65–87 |
55 |
|
|
| α-hemolysin |
88–93 |
88, 90, 94, 95 |
90, 93, 96–99 |
90, 93, 100–102 |
|
|
| K+ channels |
103–109 |
110–117 |
29, 88, 111–113, 116–197 |
107, 198–202 |
203–206 |
207–211 |
| nAChR |
|
|
212–220 |
|
|
|
| MscL/MscS |
221–228 |
229, 225 |
230–257 |
225, 258 |
222, 258 |
|
| Anion channels (VDAC, CIC) |
259 |
|
260–264 |
265–268 |
|
|
| Aquaporins |
|
|
269–274 |
|
|
|
| NH4+ transporter |
|
|
275–278 |
|
|
|
| Other channels |
279–310 |
311–318 |
299, 312, 319–348 |
302, 330, 337, 349–356 |
357, 358 |
|
| Synthetic nanopores |
359–370 |
371–374 |
375–391 |
350, 382, 383, 392 |
393 |
394 |
A two-decade-old review provided analysis combining MD simulations and various associated calculations with modeling to provide approaches that help understand the structure/function relationships for channels in human cells
[9]. Here the modeling techniques were analyzed for potassium channels, the voltage-gated (Kv), and the inward rectifier (Kir) channels. The NMR structures of (the pore-lining) M2 helix were the basis on which the transmembrane region of the pore could be modeled.
What matters to understand the ion channel function is based mostly on two things: (i) ion channel pore region geometry, and (ii) energetics that controls the pore opening/closing phenomena. Direct and indirect experimental techniques usually can address them phenomenologically but underlying mechanisms largely rely on modeling of the channel using bioinformatics techniques
[8].
Taking the potassium channel as an example case, Heil and colleagues introduced an interesting bioinformatics method, the so-called ‘Property Signature Method’ (PSM), to address this issue of identification of the channel sequences
[10]. This technique relies on physicochemical amino acid properties, instead of amino acid building blocks. A pore region signature (including the selectivity filter) was created, representing the most common physicochemical properties of the known potassium channel, thus enabling the genome-wide screening for the sequences having similar features, despite having low degree of the amino acid similarity within any specific family of the protein.
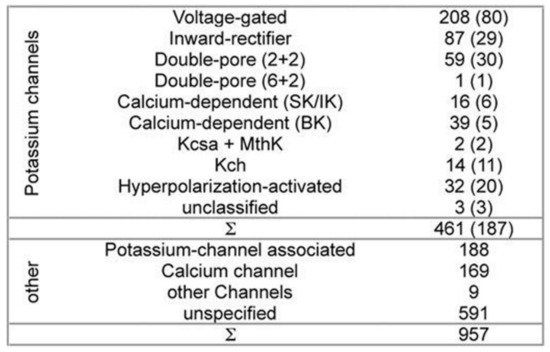
While developing PSM the dataset used 461 potassium channel α-subunits that represent different family types, see
Figure 1 [10]. A pairwise similarity of the sequences <80% were considered (187 sequences). The set was considered to contain additional 957 non-α-subunits, so that false positive could be provided. The sequences included ion channels that are closely related. All of the sequences used here have been extracted from the Swiss-Prot
[11].
Figure 1. Channel families-the composition of dataset. All of the sequences have been extracted from the Swiss-Prot
[11]. Potassium channels represent both the different families and the topologies of the known channels. Non-potassium channels here have been used as the false positives. All of the sequences having >80% sequence-similarity have been removed. The remaining channel numbers are in brackets. The (2 + 2) channels’ double-pore consists of 2 α-subunits having 4 transmembrane domains each. The α-subunits of the (6 + 2) channels with double-pore possess 8 transmembrane domains. Ambiguity exists in 3 unclassified potassium-channels classification.
The pore region profile for a potassium channel was created with the use of the dataset. The profile wasn’t used for describing the conserved positions of the amino acids in the region. But it described all of the variations in various families of the potassium channels. The profile then was translated into creating a descriptor, which describes various sequence region properties. Each profile position located amino acids got analyzed, and the properties with conserved absence or presence were used in order to describe the mentioned position. Here the Hits were ranked following the properties that were found in the property descriptor and in the target sequence. The algorithm of screening was created in the C++ language of programming.
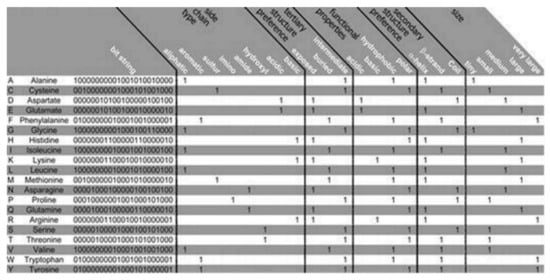
The PSM is found to use the representation of the amino acid via consideration of a binary signature that was derived using varieties of physicochemical properties. Altogether, 23 properties were used, combined into 5 groups as follows: the side chain type, the functional properties, the secondary and the tertiary structure (preference), and size, see
Figure 2 [10]. Each amino acid is represented by a created binary string, where a bit has been set to 1 for a corresponding property found to apply to the considered amino acid. Five bits have been set, one for every group of the property. Zero is assigned for all of the bits that are remaining. Thus, 20-bit strings (unique) have been found, 1 for every amino acid, which was used in this algorithm. Two steps are considered in the method as follows:
Figure 2. Amino acid properties. The Bit string that represents the amino acids with 23 properties has been presented. The relative occurance frequency got converted into corresponding binary values with the aid of the majority vote. Regarding ‘size’ all of the amino acids got categorized considering the molecular weights: tiny, small, medium, large, and very large for ≤71 Da, ≤103 Da, ≤115 Da, ≤137 Da, and >137 Da, respectively.
- (i)
-
an aligned pore domain profile was created including all of the amino acids that were present (in >3% of the investigated total 461 potassium channels),
- (ii)
-
the profile was translated into a representing string consisting of the sequences’ physicochemical properties.
Large-scale potassium channel sequence analysis confirms the requirement of identifying the potassium channel α-subunit proteins
[12]. As the family of the potassium channel is found to be highly diverse and also closely related to many other ion channels, the use of the amino acids in order to classify the potassium channels in PSM has been found imprecise. PSM is found superior over Markov models and the BLASTp, see refs.
[12][13][14]. Moreover, the PRINTS Database provided potassium channel motifs are used
[15]. These approaches are found to utilize multiple methods to overcome a method’s limitations of recognizing the potassium channel family’s subset sequences. These issues are indeed resolved in PSM. Because it can detect properties representing all of the subsets of the family of the potassium channels. Moreover, PSM is able to analyze amino acids’ physicochemically relevant properties and enables pretty sensitive extraction of the information that is coded in the sequences of the amino acids. For details, readers may consult the original article
[10].
The
Saccharomyces cerevisiae genome was well screened applying PSM
[10]. Two hits were found, the domains in the pore in the two-pore potassium channel, TOK1, which is the only one known as the S.cerevisiae potassium channel. Despite having a strong relationship including high homology among the potassium transporters, TRK1 and TRK2, to the potassium selective domains of the pore of TOK1, the mentioned two are classified as nothing but the non-potassium channels.
Heil and colleaques also performed another test with Caenorhabditis elegans having a complete genome sequence
[16]. Its genome regarding the sequences of the potassium channels is well understood; almost 40 double-pore domains have been annotated. PSM helped recover all. Additionally, a new (potential) pore domain was identified.
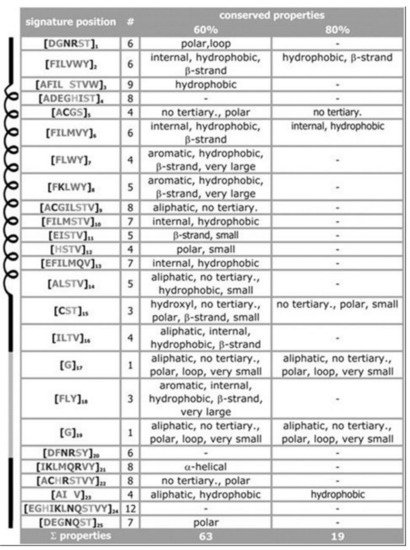
For the signature of the potassium channel, a summary of the conserved properties (at 60% with 80% threshold of conservation) is presented, see Figure 3. Despite considerable sequence set divergency, as many as 63 properties are found conserved with as high as 60% level of significance, and 19 properties are found conserved with as high as 80% level of significance. Unusual properties (not shown) coded in signature; almost 350 properties with 60% level of significance and 330 properties with 80% level of significance. The method specificity draws significant contributions from these mentioned properties.
Figure 3. Property conservation at the 60 and 80% level of significance, respectively. Despite having low amount of amino acid conservations we find properties conserved in almost 80% sequences. From pores of the potassium channels, as expected, the hydrophobic residues are found to dominate in pore regions, a few polar residues decrease energetic barriers for K+ ions. Details in ref.
[10].
PSM is considered superior to other conventional methods while searching for the sequences having a pretty low level of conservation. PSM has an important advantage. For every amino acid position, the signature describes the frequent properties (selected and uncommon ones) in the α-subunit portion of the potassium channel. The use of the position-bound signature properties has additional advantages, interpretation of the results appears pretty simple. Next to the missing and unusual number properties, this method is found to return, for every sequence, the display of a vector whose sequence positions are found to contain the untypical and missing residues, respectively, thus facilitating the fast sequence analysis.
2. Ion channel Genomes Track the Early Animal Evolution
A comparative study of genomics provides novel windows into the (confusing) past that may be applied for the understanding of the early nervous systems evolution of the animal kingdom
[17]. There is a controversy on nervous systems whether they got evolved just once, or independently being distinctive in various animal lineages. Liebeskind and colleagues explored the historical aspects of the gene families of the ion channels, central to the function of the nervous system. They tracked the timeline when the families of the genes expanded in the evolution of the animal and discovered the gene families to be radiated on multiple occasions, occasionally, they underwent various periods of contraction. Multiple gene family origins may be considered to signify considerably the large-scale evolution convergence for the complexity of the nervous system.
The ancestral gene content reconstruction helped was used in tracking the gene family’s expansion timing. Here the majority of the ion-channel protein families that may drive nervous system functions are used. Animals having nervous systems are found broadly to have identical complements of the types of ion channels. But it was also found that these complements could have been evolved independently. Ion channel gene family evolution was found to experience a large amount of loss events, among those two were found to immediately be followed by a few rounds of duplications. Ctenophores, cnidarians, and bilaterians have been found to undergo independent bouts of the gene expansion in the involved channel families connected to the synaptic transmission and the shaping of the action potential, suggesting the genomic signature of the expanding complexity in the nervous system. Ancestral nodes, where the nervous systems probably originated, were found to experience not-so-large expansions. This suggests for the origin of nerves not to experience any immediate complexity bursts, instead, the complexity of evolution perhaps experienced a rather slow fuse in the stem animals, which got followed by gene gains and losses independently.
A custom bioinformatics pipeline
[17] was used for collecting and annotating proteins that are predicted in a group of 16 families of ion channels, see
Table 2 where 41 sample opisthokonts (this group includes animal, fungi, and related protest members), and an apusozoan outgroup are presented. The channels’ families are found to be playing diversified roles in the nervous systems. Some families (e.g., the families of the voltage-gated ion channels) are found to solely be associated with the function of the nervous systems in the animals, while others (e.g., P2X receptors) are found to play relatively diverse types of roles. Only a handful of isoforms are expressed in the nervous systems. The dataset then got used in order to infer the ancestral contents of the genome and understand the timing of the happening of the gene duplications with the help of EvolMap
[18].
Table 2. Ion channel families
[17].
| Abbreviation |
Full Names |
Function |
| Ano |
Anoctamin, Ca2+ activated Cl− |
Smooth muscle, excitability |
| ASC |
Epithelial (ENaC), acid sensing channel (ASIC) |
Osmoregulation, synaptic transmission |
| CNG/HCN |
Cyc. nucleotide gated |
Sensory transduction, heart |
| Cav |
Voltage-gated Ca+ channel |
AP, muscle contraction, secretion |
| ClC |
Voltage-gated Cl− channel |
Muscle membrane potential, kidney |
| GIC |
Glutamate receptor (iGluR) |
Synaptic transmission |
| LIC |
Ligand-gated, Cys-loop receptor |
Synaptic transmission |
| Kv |
Voltage-gated K+ channel |
AP, membrane potential regulation |
| Nav |
Voltage-gated Na+ channel |
AP propagation |
| Leak |
Sodium leak (NALCN), yeast calcium channel (Cch1) |
Regulation of excitability |
| P2X |
Purinurgic receptor |
Vascular tone, swelling |
| PCC |
Polycystine, Mucolipin |
Sensory transduction, kidney |
| RyR |
Ryanodine receptor, IP3 receptor |
Intracellular, muscle contraction |
| Slo |
Voltage and ligand-gated K+ |
AP, resting potential |
| TPC |
Two-pore channel |
Intracellular, NAADP signaling |
| TRP |
Transient receptor potential |
Sensory transduction |
These gene families were found ancient
[19][20]. All except for two, acid-sensing channel (ASC) and the Cys-loop receptor (LIC), are found in the most recent common ancestor (MRCA) of the examined taxa
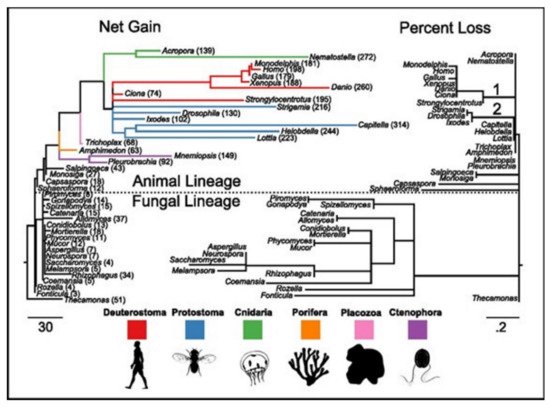
[17]. ASC family was the only one found as the metazoan-specific. The families were pulled together and they then plotted the net gains and the percent losses (on the species tree), see
Figure 4 [17]. The animal lineage was dominated by the gains but losses led to the fungal lineage.
Figure 4. The families of the ion-channels in opisthokont evolution. Both trees contain similar topologies. The lengths of the branch of the left tree are actually net gain, gains-losses. The lengths of the branch of the right tree are the percent loss, losses-gains as a% of the parent copy number. Total ion channel numbers in every taxon are presented on the left tree. Two animal branches having large loss events have been labeled-the common deuterostome and ecdysozoan ancestors.
In phylogenetic gain and loss patterns for all of 16 families of ion channels (
Table 2), large expansions of LIC, voltage-gated potassium channel (Kv), and glutamate-gated channel (GIC) families at multiple places were reported, see details in ref.
[17]. This independent gene-(family) expansions lead to MRCAs of the bilaterians, the vertebrates, and the cnidarians
[20].
Ecdysozoans and lophotrocozoans were found to have large expansions in LIC, GIC, and Kv channels. A huge expansion in ASC family was also observed, see
Figure 5A
[17]. These expansions were observed to have happened in terminal lineages that led to every species, see
Figure 5.
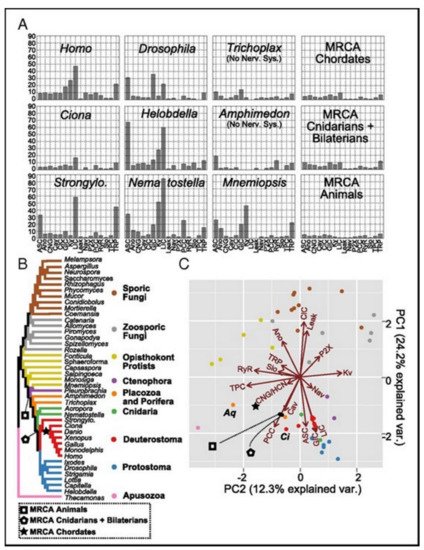
Figure 5A presents the family count of ion channels from species of the major lineage. All taxa with nervous systems, with the notable exception of the tunicate Ciona, were enriched for similar gene families. Two taxa (without the nervous systems), Trichoplax and Amphimedon, were found to have smaller complements of ion channels. MRCAs of the chordates, the cnidarians plus the bilaterians, and the animals each were found to have ion-channel complements resembling the extant animals having no nervous systems more than animals having nervous systems.
Figure 5. (A) The count of the channels of the extant and the ancestral species. (B) The species tree shows the relationships between the extant taxa and key ancestral nodes’ locations. (C) The PCA of the normalized gene contents of the ion channels for all tips and three ancestral nodes. The proximity in the space of two PCs indicates identical contents of the gene. The ion-channel families loadings have been presented as vectors. The loading vector size and direction indicate its correlation with corresponding two components. The loading arrows are pointing to the regions where the gene family is found in high relative abundance. The labeled species represent Amphimedon (Aq), and Ciona (Ci).
3. Bioinformatics Prediction of Ion Channel Genes and Channel Classification
Ion channels are indirectly or directly associated with various types of cellular disorders leading to specific diseases. Ion channels are therefore therapeutic and diagnostic targets of many drugs. About 700 drugs are known so far to act upon ion channels
[21]. Knowledge of ion channel genes and their mutations certainly is key to understanding diseases and planning for drug discovery. Bioinformatics techniques may be found quite helpful in understanding the roles of ion channels in diseases through analysis of genetics-based classifications
[21], as well as genetic mutations
[22][23] of ion channels. AI techniques have been found to play important roles in both predicting ion channel genes and understanding genetic mutations and connecting them with classified diseases. We wish to elaborate these features quite in detail here.
AI Techniques Help Predicting Ion Channel Genes
ML, a subset of AI, was used recently to extract the feature vectors of various ion channels
[21]. The SVMProt and the k-skip-n-gram methods were used, which helped obtain 188- and 400-dimensional features, respectively. SVMProt, a web-based support vector machine software, was developed for mainly functional classification of any protein considering its primary sequence
[24]. In the case where the structural protein class is inconsiderably correlated with its constituent amino acids, the support vector machine appeared as a computational tool that could predict the structural protein classes
[25]. In the k-skip-n-gram method every protein sequence needs to be transferred into a vector. Then the training vectors are used for training the random forest parameters. The testing vectors evaluate the method’s performance.
Various bioinformatics softwares are available to predict the ion channel identifications in membranes. A series of high-throughput computational tools are now available which help predict not only the ion channels but also their types directly using the protein sequences, helping in ion channel targeted drug discovery research. During last decade, many ML algorithm-based computational methods have been developed
[26][27], which may be used in drug repositioning. Saha and colleagues used the amino acid and the dipeptide compositions as feature vectors, then classified them with the use of a support vector machine (SVM) so that they could predict the voltage-gated ion channels, and their available subtypes
[28]. The identification method for a voltage-gated potassium channel, based on the local sequence information, was also proposed later by another group
[29]. The latter is found better than that developed for the identification of the voltage-gated potassium channels, based on the global sequence information
[30]. A support vector machine (SVM)-based model was recently constructed which helps predict quickly
[31]. A SVM-based model to search the predicted ion channels and subfamilies that uses the sequence similarity search features of the basic local alignment search tools was developed recently
[32].
In a recent article, the application of ML Methods in ion channels has been briefed
[33]. The review focusses on prediction methods developments for ion channels considering a few issues as follows:
-
ion channel proteins datasets,
-
predicting ion channels using ML methods,
-
obtaining the optimal ion channel prediction features using feature selection technique,
-
the prospect of bioinformatics methods prediction of ion channels using appropriate and available tools.
Han and colleagues used SVM and random forest classifiers in order to identify first the ion channels, and further to classify them
[21]. The feature selection was made using the maximum-relevance-maximum-distance (MRMD) method that helped improve the accuracy of the prediction. Three steps were followed. Firstly, a protein sequence got detected to check if it might belong to any ion channel. If the positive, then the sequence of the protein got classified as to belong to voltage-gated or ligand-gated ion channels. Finally, if the sequence belonged to the voltage-gated ion channel family, the classification was made regarding them to belong to the potassium (K
+), the sodium (Na
+), the calcium (Ca
2+), or the anion voltage-gated ion channel class.
The flowchart shows the stepwise adopted basic processes that Han and colleagues considered for the gene detection and the channel classification, see
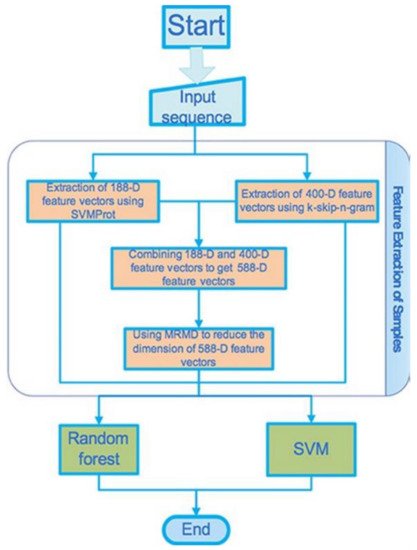
Figure 6 [21]. We avoid explaining how they introduce the set of data, the method of the feature extraction, the method of the dimension reduction, and the classifier that were used in the study, but the readers may find them in the original article.
Figure 6. Flowchart representing the proposed processes.
The original data used for the prediction model can be found in ref.
[30]. The ion channel sequences have been collected from the depository Universal Protein Resource (UniProt) and the depository Ligand-Gated Ion channel databases
[33][34]. The total number of the voltage-gated ion channels was 148; 81, 29, 12, 26 of them are potassium channels, calcium channels, sodium channels, the anion channels, respectively. Finally, 150 ligand-gated ion channels were extracted. From the UniProt 300, protein sequences were selected randomly as the non-ion channels, having the consistency of these non-ion channel sequences < 40%. Two ML methods (feature extraction methods), SVMProt 188-D relying on the protein composition and the physicochemical properties, and k-skip-n-gram 400-D were used. These two (feature representation) methods were then combined in order to form a new feature vector that contains multiple (more than one) features. The new feature vector set was then classified using the SVM and the random forest classifiers. MRMD based dimensionality reduction method (see the site
http://lab.malab.cn/soft/MRMD/index_en.html, updated by Prof. Quan Zou on 2 November 2016) was then employed for reducing the generated feature vectors’ dimensionality
[35]. The MRMD works in selecting the feature having the highest correlation and the least redundancy through calculation of the maximum distance and relevance. Here, they used a random forest classifier for building the model. As this classifier uses multiple trees for training and predicting samples, this one is popularly used in bioinformatics research where applicable, e.g., see ref.
[36]. It is found a good performing tool, using especially the random forest algorithm
[37], in many practically relevant fields, e.g., the regression and classification of the gene sequences, the action recognition, the face recognition, the anomaly detection in data mining, and the metric learning.
The effects of the prediction of the random forest-based and SVM-based methods on both non-ion and ion channels in various dimensions were compared in this study, see the results in
Table 3 [21]. The results for 10-fold cross-validations of 188- and 400-dimensional features and their mixed features have been listed in
Table 3. The MRMD method was then applied to reduce 27 dimensions from 588-dimensional features for obtaining 587-dimensional features, with the latter having average classification accuracy lower than that found for the 400-dimensional features. The SVM classifier was reported to be the best to classify the 400-dimensional features. The average overall accuracy (OA) rate, 85.1%. 86.6% of the ion channels, and 83.7% of the non-ion channels, can be identified approximately by the SVM classifier. A total 85.1% accuracy was obtained. Thus feature vectors from 188- and 400- dimensional features yield pretty acceptable prediction results.
Table 3. Prediction results of the ion channels and the non-ion channels.
| Method |
Ion Channel (%) |
Non-Ion Channel (%) |
OA (%) |
| Random forest (188D) |
90.3 |
77.2 |
83.7793 |
| SVM (188D) |
87.0 |
78.5 |
82.7759 |
| Random forest (400D) |
87.7 |
77.5 |
82.6087 |
| SVM (400D) |
86.6 |
83.7 |
85.1171 |
| Random forest (588D) |
77.5 |
90 |
83.7793 |
| SVM (588D) |
83.2 |
80 |
81.6054 |
| Random forest (587D) |
77.2 |
89.7 |
83.4448 |
| SVM (587D) |
77.2 |
83.3 |
80.2676 |
The accuracy was evaluated on 188-, 400-dimensional features, and their mixed features, and 88-dimensional features that were obtained following dimensional reduction with the use of MRMD which discriminates between classification results of the voltage-gated and the ligand-gated channels. All these results are summarized in
Table 4 for these two classes and in
Table 5 for ion specificity in voltage-gated ion channels
[21]. 93.9% and 86.0% of the voltage-gated and ligand-gated ion channels, respectively, could correctly be identified with the use of the random forest method. This classifier is a better performer than the SVM classifier especially in a few cases, and also can provide an improved prediction performance model.
Table 4. Compare the results of the voltage-gated ion channels with that of the ligand-gated ion channels.
| Method |
Voltage-Gated
Ion Channel (%) |
Ligand-Gated
Ion Channel (%) |
OA (%) |
| Random forest (188D) |
93.9 |
86.0 |
89.9329 |
| SVM (188D) |
91.9 |
86.7 |
89.2617 |
| Random forest (400D) |
88.5 |
82.7 |
85.5705 |
| SVM (400D) |
82.4 |
83.3 |
82.8859 |
| Random forest (588D) |
89.2 |
86.0 |
87.5839 |
| SVM (588D) |
91.9 |
86.7 |
89.2617 |
| Random forest (188D) |
92.6 |
86.7 |
89.5973 |
| SVM (188D) |
91.9 |
86.7 |
89.2617 |
Table 5. Prediction results for the voltage-gated ion channels-four types.
| Method |
K (%) |
Ca (%) |
Na (%) |
Anion (%) |
OA (%) |
AA (%) |
| Random forest (188D) |
97.5 |
37.9 |
50 |
46.2 |
72.973 |
57.9 |
| SVM (188D) |
96.3 |
48.3 |
58.3 |
69.2 |
79.0541 |
68.0 |
| Random forest (400D) |
97.5 |
6.9 |
50 |
23.1 |
62.8378 |
44.4 |
| SVM (400D) |
85.2 |
62.1 |
50 |
73.1 |
75.6757 |
67.6 |
| Random forest (588D) |
97.5 |
34.5 |
50 |
57.7 |
74.3243 |
59.9 |
| SVM (588D) |
96.3 |
48.3 |
58.3 |
69.2 |
79.0541 |
60.2 |
| Random forest (424D) |
98.8 |
34.5 |
58.3 |
46.2 |
73.6486 |
59.5 |
| SVM (424D) |
96.3 |
48.3 |
58.3 |
69.2 |
79.0541 |
68.0 |
 +1 credit
+1 credit