Diseases primarily affecting the basal ganglia in children result in characteristic disturbances of movement and muscle tone. Both experimental and clinical evidence indicates that the basal ganglia also play a role in higher mental states. The basal ganglia can be affected by neurometabolic, degenerative diseases or other conditions from which they must be differentiated. Neuroradiological findings in basal ganglia diseases are also known. However, they may be similar in different diseases. Their assessment in children may require repeated MRI examinations depending on the stage of brain development (mainly the level of myelination). A large spectrum of pathological changes in the basal ganglia in many diseases is caused by their vulnerability to metabolic abnormalities and chemical or ischemic trauma. The diagnosis is usually established by correlation of clinical and radiological findings. Neuroimaging of basal ganglia in neurometabolic diseases is helpful in early diagnosis and monitoring of changes for optimal therapy.
- basal ganglia abnormalities
- children
- neurometabolic disease
1. Introduction
Anatomically, the basal ganglia are deep symmetrical structures of the gray matter. Most authors consider the following structures to be part of the basal ganglia: caudate and lentiform nuclei, thalamus, subthalamic nuclei and substantia nigra [1]. The lentiform nucleus consists of the inner globus pallidus and outer putamen (putamen belongs to the structure of the striatum together with a distinct caudate nucleus) (Figure 1).
The basal ganglia are involved in both motor and non-motor functions, including higher order cognition, social interactions, speech and repetitive behaviors [2–6]. The first and main clinical symptoms of diseases with basal ganglia involvement are disorders of movement and muscle tone [1].
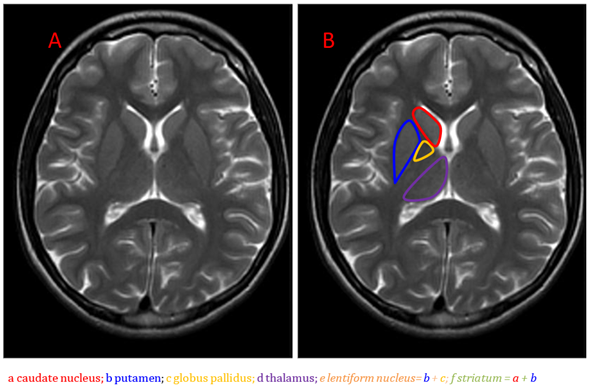
Figure 1. Brain anatomy on MRI, the basal ganglia on the T2-weighted image, axial plane (A,B).
Due to subtle differences between the white and gray matter, magnetic resonance is the imaging method of choice because density changes on computed tomography (CT) scans are mostly unnoticed. Imaging of the basal ganglia in children is challenging due to infantile and childhood brain development and significant physiological changes in the appearance and signal of brain tissues.
In neonates, the white matter is iso- to hypointense compared to the gray matter on T1-weighted images (T1-WI) and iso- to hyperintense on T2-weighted images (T2-WI). Therefore, it is difficult to differentiate between the white and gray matter in the basal ganglia due to the lack of myelination in the internal capsule (Figure 2). Signal intensity at the posterior limb is changed, which results in a consecutive increase in T1 signal intensity (at 0–1 month) and a decrease in T2 signal intensity (at 0–3 months) [2] (Figure 3). Myelination of the anterior limb is a longer process (increase in signal intensity on T1-WI between the 2nd and 3rd month and decrease on T2-WI between the 7th and 11th month) [2] (Figure 4).
Location of changes in the deep nuclei depends on different degrees of sensitivity to different insults.
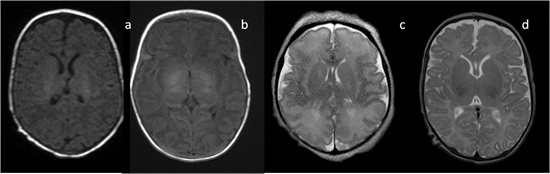
Figure 2. Infant brain MRI—no differences in signal intensity between the basal ganglia and the surrounding structures before the completion of myelination: (a,b) axial T1-weighted images (T1-WI) in a neonate with a hyperintense signal in the posterior internal capsule; (c) axial T2-weighted images (T2-WI) at the 36th week of gestation; (d) axial T2-WI in a neonate.
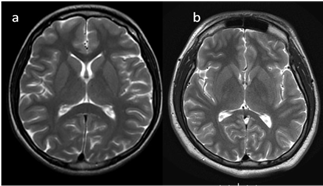
Figure 3. (a,b). Completion of myelination on brain MRI and differences in signal intensity between the basal ganglia and the surrounding brain tissues (axial T2-WI).
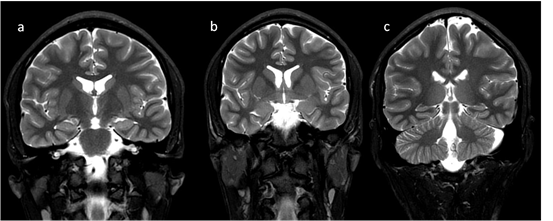
Figure 4. Completion of myelination on brain MRI and differences in signal intensity between the basal ganglia and the surrounding brain—coronal T2-WI; putamen and caudate nucleus head (a,b); thalamus and caudate body (c).
Many diseases, including some neurometabolic disorders, change signal intensity from the basal ganglia. Their assessment may require repeated examinations depending on the stage of brain development (mainly the level of myelination). Basal ganglia imaging consists of multiple sequences including T1-WI, T2-WI, T2 * GRE/SWI, diffusion-weighted imaging (DWI) and contrast-enhanced imaging. These options are commonly used in clinical practice.
Changes in the basal ganglia signal might be of hyper-, hypo- or mixed intensity on T1-WI, hyper-, hypo- or mixed intensity on T2-WI or MRI-related artefacts (hemoglobin, calcium, iron, DWI) with the presence or the lack of gadolinium contrast enhancement. Of note, the imaging studies provide the description of the distribution of pathology and symmetry or asymmetry of lesions [7]. Advanced imaging methods such as positron emission tomography (PET), diffusion tensor imaging (DTI), perfusion-weighted imaging (PWI) and MR spectroscopy (MRS) provide a deeper insight into basal ganglia with an additional analysis of the metabolites. Therefore, PET examination is more useful in the assessment of adults, mainly due to its high radiation dose. Pediatric patients can benefit from safer and more advanced MRI techniques. Diffusion tensor tractography (DTI) provides more precise information on motor disorders. Some diseases might cause both hyper- and hypointensity, e.g., “the eye of the tiger” sign or the Parkinson variant [7–9].
In clinical practice, MRI of the head is used as the initial examination of all patients suspected of having neurometabolic diseases with the basic protocol containing the sequences T1-WI, T2-WI, T2 * GRE/SWI, diffusion-weighted imaging (DWI). In some patients, MRS examination, due to a detailed analysis of the presence of metabolites typical for a given disease, allows for its confirmation or exclusion. DTI analysis is rarely used—more often in research work than in everyday practice. In the diagnosis of neurometabolic diseases in children, the use of PET as a method that exposes a high dose of radiation is avoided.
Precise identification of calcium, hemosiderin, iron or metals is not always possible and might be easier with special sequences (e.g., T2 * GRE).
A large spectrum of pathological changes in the basal ganglia in many diseases is caused by their vulnerability to metabolic abnormalities [7,8]. Physiological high activity with high glucose and oxygen uptake, rich vascular supply, a large amount of mitochondria and chemical substances make the basal ganglia very sensitive to metabolic, chemical or ischemic trauma [8].
Most metabolic diseases and some vascular disorders (e.g., central venous infarction) cause symmetrical basal ganglia changes [7,9]. Asymmetry of the pathology usually leads to the diagnosis of infarction or ischemia, an infectious or neoplastic process, which are not discussed in this paper as they are beyond the scope of the article [7,8]. Location of abnormal signal intensity in the basal ganglia (Table 1) aids in the differential diagnosis [7–9]. Contrast administration is useful in infarction, encephalitis and neoplastic conditions. Most changes in metabolic diseases are not enhanced following gadolinium administration. The use of advanced techniques (PWI, DTI, MRS) renders MRI a powerful tool.
Table 1. Abnormal MR signal characteristics in particular sequences (T2-WI, T1-WI, DWI); metabolic diseases are given in bold [7–9].
|
Basal Ganglia MRI Signal Intensity |
Disease |
|
T2 hyper = increased signal on T2-WI |
Hypoxic-ischemic encephalopathy (early) CO toxicity Kernicterus Mitochondrial diseases (e.g., Leigh disease) Infantile bilateral striatal necrosis Hypoglycemia Infarction Rasmussen encephalitis NF1 Myelinolysis Intoxications |
|
T2 hypo = decreased signal on T2-WI |
(acute) Deoxyhemoglobin Hypoxia |
|
T1 hyper = increased signal on T1-WI |
Gadolinium concentration after chelate administration Hypoxic-ischemic encephalopathy (late) CO toxicity NF1 Manganese toxicity (subacute) Methemoglobin Liver failure |
|
T1 hypo = decreased signal on T1-WI |
Infarction Infantile bilateral striatal necrosis |
|
DWI restriction = increased signal on DWI with corresponding decreased signal on apparent diffusion coefficient (ADC) maps |
Infarction Hypoxic-ischemic encephalopathy CO toxicity Mitochondrial diseases Encephalitis of different origin Wilson disease |
T2-WI: T2-weighted images, T1-WI: T1-weighted images, DWI: diffusion-weighted imaging.
2. Neurometabolic Disorders
2.1. Mitochondrial Disorders
The prevalence of metabolic stroke in myoclonic epilepsy with ragged red fibers (MERRF) is even 60%, whereas in mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS), it is 84–99%. Even in 13% of children, MELAS and MERRF have been associated with calcification of the basal ganglia [11,12].
In the literature, the neuroimaging spectrum ranges from classical Leigh syndrome with symmetrical lesions in the basal ganglia and/or brainstem to structural abnormalities including cerebellar hypoplasia and corpus callosum dysgenesis [11–13]. Due to advances in genomic technology, mainly in next-generation sequencing (NGS) and whole-exome sequencing (WES), the genetic spectrum of Leigh syndrome has broadened and, so far, 75 genes have been identified both in mitochondrial (30%) and nuclear DNA [11–13]. Leigh syndrome is one of the most common mitochondrial disorders.
In the rare mitochondrial encephalomyelopathy (Alpers disease), a “pulvinar sign”—bilateral symmetric pulvinar (mediodorsal thalamic nucleus) T2 high signal relative to the signal intensity of other deep gray matter nuclei and cortical—has been described [11–13]. This symptom is non-specific; it may occur in other diseases such as Creutzfeldt–Jacobs disease (CJD), postinfectious encephalitis and cat-scratch disease [12,13]. Differentiation is based on clinical data, including age, and other underlying lesions in the brain.
Stroke-like episodes or metabolic strokes are related to neurological deficits due to biochemical abnormalities in focal brain regions that are not connected with ischemic or hemorrhagic causes. Apart from mitochondrial defects, stroke-like episodes also occur in aminoacidopathies, urea cycle defects, lysosomal storage disorders or organic acidemias [14,15]. Almuqbil et al. reported on a patient with propionic aciduria with basal ganglia changes on brain MRI several years after the resolution of neurological symptoms [15]. MRI, angio-MRI and MRI spectroscopy are useful tools in differentiating between vascular and nonvascular factors.
2.2. Neurodegeneration with Brain Iron Accumulation (NBIA)
This group of diseases is characterized by abnormal iron accumulation in the basal ganglia, leading to different progressive movement and developmental disorders. The subtypes of Neurodegeneration with Brain Iron Accumulation (NBIA) are connected to specific gene disorders. There are four main types including pantothenate kinase-associated neurodegeneration (PKAN), PLA 2G6-associated neurodegeneration (PLAN), mitochondrial-membrane protein associated neurodegeneration (MPAN) and beta-propeller protein associated neurodegeneration (BPAN). There are also other rare, familial forms of NBIA [16].
The radiological pattern in NBIA is described as the “eye of the tiger sign”. Iron storage is evident in the anteromedial part of the globus pallidus as central hyperintensity on T2-WI with the surrounding area of hypointensity [8].
Iron deposits may be also observed in Parkinson disease, atypical parkinsonian disorders, multiple sclerosis or Friedreich ataxia [10]. The presence of iron deposits in the basal ganglia is age-dependent and is not found at birth, which is crucial for the differential diagnosis [10]. MRI changes may precede clinical symptoms.
2.3. Biotin-Thiamine-Responsive Basal Ganglia Disease (BTBGD)
Biotin-thiamine-responsive basal ganglia disease (BTBGD), also known as thiamine transporter-2 deficiency, is an autosomal recessive disorder caused by SLC19A3 gene mutation [17]. In BTBGD, episodes of recurrent encephalopathy (confusion, seizures, dystonia, ataxia, dysphagia, supranuclear facial palsy, external ophthalmoplegia) are triggered by febrile illness or stress [17,18]. Neuroradiologically, it is characterized by bilateral necrosis and severe edema within the basal ganglia in acute stages. The most typical MRI abnormalities include central destruction of the caudate heads and partial or complete loss of the putamen with diffusion restriction and sparing of the globus pallidus [16,17]. Alfhadel et al. reported diffuse cortical, subcortical and white matter involvement in acute phase next to basal ganglia pathologies on 18 patients [19,20]. Similarly, Kaseem et al. in their study on 15 patients observed coexisting changes in the mesencephalon, cerebral cortical and subcortical regions and thalamus in 80% of cases [21]. In 53%, patchy deep white matter affection was found, and the cerebellum was involved in 13.3% [21]. After thiamine and biotin therapy, resolution of changes was found in the mesencephalon, white matter and cortex. Kamasak et al. confirmed a typical involvement of the putamen and caudate with diffusion restriction [22]. In BTBGD, treatment with high doses of biotin and thiamine may influence the disease progression and seizure activity [22].
2.4. Pyruvate Dehydrogenase Deficiency (PDHD)
Pyruvate dehydrogenase deficiency (PDHD) results in early-onset encephalopathy with X-linked recessive inheritance. Newborns usually present with severe lactic acidosis and subsequently develop significant neurological defects similar to those observed in patients with Leigh syndrome [10–12]. Children with later-onset forms may present with paroxysmal neurological episodes, often precipitated by fever or exercise and frequently associated with psychomotor retardation and pyramidal tract signs [23]. Brain MRI mostly shows bilateral cystic lesions of the putamen and the brain parenchyma, coexisting with corpus callosum abnormalities, asymmetric hydrocephalus and small pons [23]. In less severe forms, basal ganglia lesions seen on MRI might be reversible with treatment, as shown by follow-up MRI.
2.5. Glutaric Aciduria (GA) Type 1
Typical cases are characterized by early progressive macrocephaly and/or hypotonia. Although the imaging pattern is not characteristic, widening of the Sylvian fissures with associated macrocephaly should suggest searching for GM1 [24–26]. Additional changes in the striatum (putamen and caudate) were typically found in the acute stage. The globus pallidus is affected less frequently than striatum. However, changes in cerebellar dentate nuclei may be present, as in the case of L-2-hydroxyglutaric aciduria [24–26].
2.6. Gangliosidosis (GM1 and GM2)
All gangliosidoses affect the volume of the cerebrum and cerebellum. Movement disorders, including tremor, generalized or focal dystonia, chorea and parkinsonism, are present in 30–50% of late-onset (juvenile and adult) forms, and more volumetric changes of brain structures in these forms were reported, with thalamic hyperintensities (infantile form) [7–10].
Infantile (type 1) and juvenile (type 2) forms cause signal abnormalities of the basal ganglia [10,27]:
- in GM1, hyperintensities of posterior putamen and globi pallidi on T1-WI with susceptibility effects on T2-WI were found;
- in GM2, thalamic hyperintensity on T1-WI, mixed striatum signal intensity on T2-WI, hypointense white matter on T2-WI (Tay–Sachs syndrome: hypointense on T2-WI (ventral thalamus) hyperintense on T2-WI (dorsal thalamus); Sandhoff disease: T2 hypointense thalami).
Changes in deep nuclei are more typical of infantile GM2, whereas juvenile and adult forms present with cerebellar hypo/atrophy.
2.7. Wilson Disease
Wilson disease, the effect of ceruloplasmin deficiency, results in copper storage in the liver and subsequentially in the thalamus, putamen and brain stem. The abnormal signal at the level of brain stem is referred to as the “giant panda face” (on T2-WI: symmetrical hyperintensity in tegmentum, and comparatively low-preserved signal in red nuclei, pars reticularis and superior colliculi) and/or “miniature panda face” (on T2-WI: hyperintense aqueduct entrance with relatively hypointense central tegmental tracts) [28,29].
2.8. In Methylmalonic Aciduria (MMA)
An increasing signal intensity on MRI is observed, typically with selective necrosis. A better insight into the basal ganglia pathology is provided by MRS. The NAA/Cr and NAA/Cho ratios in methylmalonic aciduria are lower than in healthy children [14,30].
2.9. Fabry Disease
Fabry disease can manifest radiologically as a vast range of unspecific changes—hence the need for close clinical diagnostic workup. T1 hyperintensity of the lateral pulvinaris is thought to be pathognomonic [31].
2.10. Canavan Disease (CD)
Deep nuclei (globi pallidi and thalami) are typically affected by Canavan disease (CD), whereas the striatum and dentate nuclei are spared. MRI also reveals bilateral symmetric white-matter hyperintensity on T2-WI, including involvement of the subcortical arcuate fibers [32]. It can be observed in the white matter of the cerebellum and brain stem and, particularly, in the subcortical white matter [32]. The disease is the only known metabolic disorder which causes an increase in N-acetylaspartate levels on MR spectroscopy.
This entry is adapted from the peer-reviewed paper 10.3390/brainsci10110849