Pulmonary vascular remodeling is the critical structural alteration and pathological feature in pulmonary hypertension (PH) and involves changes in the intima, media and adventitia. Pulmonary vascular remodeling consists of the proliferation and phenotypic transformation of pulmonary artery endothelial cells (PAECs) and pulmonary artery smooth muscle cells (PASMCs) of the middle membranous pulmonary artery, as well as complex interactions involving external layer pulmonary artery fibroblasts (PAFs) and extracellular matrix (ECM). Inflammatory mechanisms,apoptosis and other factors in the vascular wall are influenced by different mechanisms that likely act in concert to drive disease progression. This article reviews these pathological changes and highlights some pathogenetic mechanisms involved in the remodeling process.
1. Introduction
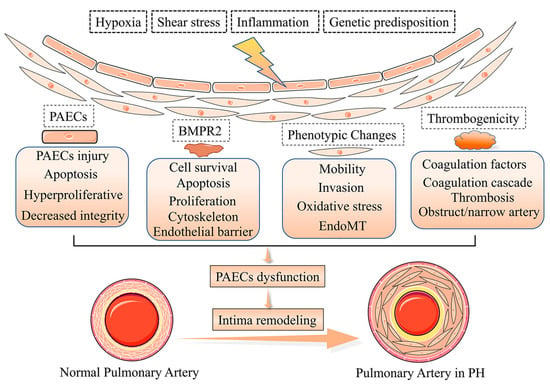
The intima represents the thick interface of endothelial cells between the media and flowing blood. The endothelial cells provide a broad unobstructed flow surface area, contributing to the suitable perfusion pressure that is the normal state of pulmonary circulation [
7]. Patients with severe PH have an approximately 3 fold increase in pulmonary intima, but the mechanism of intima damage is unclear [
8] and intimal thickening will result in an approximately 40-fold increase in resistance of the pulmonary vascular. There are various types of intimal thickening and PAECs dysfunction, as the most critical component of the intima accelerates the process of intima remodeling [
9]. Under normal physiological conditions, PAECs are in a steady state and secrete a variety of active factors that disturb PAECs and PASMCs proliferation, coagulation, the attraction of inflammatory factors and activation of vasoactivity, which leads to dysfunction and pathological changes of PASMCs in PH [
10]. Multiple factors trigger PAECs dysfunction in PH [
11,
12,
13,
14], such as shear stress, hypoxia, inflammation, PAECs phenotypes, the bone morphogenic type 2 receptor (BMPR2), and cilia length [
15,
16,
17] (
Figure 1). Meanwhile, the deterioration of endothelial metabolic function in the pulmonary vascular system is becoming an important driver of PAECs dysfunction and PH development [
18].
Figure 1. Pathogenesis of pulmonary vascular intima remodeling in PH. In response to hypoxia, shear stress, inflammation, and genetic predisposition, the function and phenotype of PACEs are altered, resulting in intima remodeling in PH.
2. Phenotypes of PAECs Dysfunction in Intima Remodeling
Damage and apoptosis of PAECs can occur in the early stages of PH pathogenesis, while anti-apoptotic PAECs appear later as PH progresses [
19]. In late PH, hyperproliferative and anti-apoptotic PAECs predominate and facilitate the formation of plexiform lesions [
20]. The pathogenesis of PH is usually associated with abnormal endothelial cell barrier integrity, and patients with idiopathic PH (iPH) often exhibit a hypercoagulable phenotype. Additionally, there is a growing awareness that complex alterations in metabolic and epigenetic pathways facilitate the progression of PH [
14]. However, it is essential to note here that PAECs include separate subpopulations of endothelial cells, which are possible exposure to multiple adverse stimuli and physical damages depending on location in the pulmonary vascular system [
21].
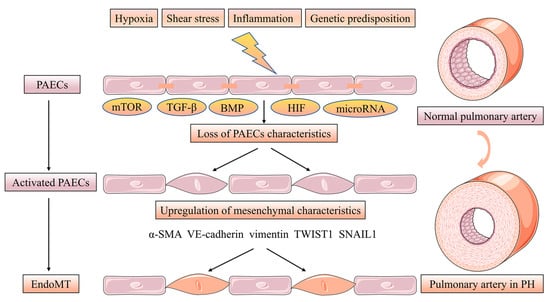
Endothelial-mesenchymal transition (EndoMT) is a phenotypic change in which PAECs manifest a mesenchymal-like phenotype with concomitant endothelial cell characteristics loss while upregulating the level of mesenchymal markers. Furthermore, PAECs adopt highly migratory and invasive cell phenotype characteristics with loss of cell-cell contact [
22]. Strongly expressed α-smooth muscle actin (α-SMA), vimentin and VE-cadherin appeared in the PAECs of human PH patients and PH rat models induced by monocrotaline (MCT) hypoxia accompanied EndoMT. PAECs treated with transforming growth factor beta (TGF-β) induce the levels of the EndoMT transcription factors TWIST1, SNAIL1 and the previously mentioned mesenchymal markers involved in this process [
23,
24]. More interestingly, BMP-7 was abrogated in hypoxia-induced PAECs by the action of EndoMT via inhibiting the mTORC1 signaling pathway [
25]. Low BMPR2 expression favors EndoMT leading to over-activated TGF-β signaling [
26]. In conclusion, altered TGF-β/BMP signaling is associated with the EndoMT process in PH [
27]. Hypoxia acts as an inducer of EndoMT via increasing hypoxia-inducible transcription factor-1α (HIF-1α) and hypoxia-inducible transcription factor-2α (HIF-2α) in PH [
28]. Finally, microRNA, such as miR-27a, miR-124 and miR-181b, can be implicated in EndoMT in PH [
29,
30,
31] (
Figure 2).
Figure 2. EndoMT of PACEs in pulmonary vascular intima remodeling. EndoMT is a phenotypic change in which PAECs manifesting a mesenchymal-like phenotype with concomitant endothelial cell characteristics loss while upregulating the level of mesenchymal markers. Furthermore, PAECs adopt highly migratory and invasive cell phenotype characteristics with losing cell-cell contact. The EndoMT process is regulated by the mTOR, TGF-β, BMP and HIF signaling pathway. microRNA, such as miR-27a, miR-124 and miR-181b can be implicated in EndoMT in PH.
3. PAECs Survival and Proliferation in Intima Remodeling
BMP receptor signaling, which is encoded by SMAD1, SMAD4 and SMAD9, plays an important role in PH development. BMPR2, as a transmembrane enzyme receptor that regulates TGF-β signaling in PAECs in the lumen of the pulmonary vessels, promotes the survival of PAECs and antagonizes PASMCs proliferation [
32,
33]. Interestingly, GDF2 encodes the circulating BMP 9, which is a ligand for the BMP2 receptor, and mutations in GDF2 reduced levels of BMP family expression [
34].
Additionally, siRNA-mediated silencing of BMPR2 in PAECs contributes to the inhibition of Ras/Raf/ERK and Ras signaling reversing proliferation and hypermotility [
35]. With the further development of pathology in PH, PAECs proliferation is a major manifestation resulting in complex arterial structural and functional remodeling, and multiple pathways regulate this transition. Peroxisome proliferator-activated receptor-γ in PAECs inhibits the cell cycle and disrupts endothelial cell barrier function, while antagonizing the migration and angiogenic properties of PAECs [
36]. Furthermore, recent studies have suggested a role for endothelial prolyl hydroxylase 2 (PHD2) in PH pathology, and mice with Tie2Core-mediated PHD2 disruption in PAECs exhibited vascular remodeling in PH [
37].
The proliferation and survival of PAECs are influenced by several other factors that also exacerbate the pathological condition of PH, such as disruption of Cav1 [
38]. mTOR, Nur77 and GDF11 also act as inhibitors of PAECs proliferation and angiogenesis after hypoxia [
39,
40]. Oxidative, antioxidant and nitrification equally affect endothelial function. Inhibition of reactive oxygen species (ROS)-induced Ca
2+ entry also downregulates the migration and proliferation of PAECs [
41]. It has been recently shown that endostatin, a cleavage product of Col18A1, inhibits PAECs proliferation and apoptosis via CD47 and ID1/TSP-1/CD36 signaling [
42]. The absence of Notch coupling to Sox17 in endothelial cells may exacerbate PH by upregulating the monolayer vulnerability of PAECs [
43]. These findings demonstrate the complex relationship between PAECs survival and proliferation in PH.
4. PAECs Activation and Thrombogenicity in Intima Remodeling
The presence of thrombotic lesions is a common pathological manifestation of PH. However, the role assumed by thrombus in PH remains controversial [
44]. Multiple factors participate in the regulation of PH by affecting PAECs activation and thrombogenicity. A few studies have shown that coagulation factors, represented by activators of the coagulation cascade, lead to the aggregation of fibrin clots and blockage of blood vessels and exacerbate PAECs dysfunction, leading to vascular remodeling [
45]. The levels of von Willebrand Factor (vWF) in PH patients also increase in the plasma. PAECs and platelets express and release vWF when activated, facilitating their interaction. The levels of thrombomodulin, as a series of anti-coagulant factors, are decreased in PH patients, which can inhibit this deterioration by ingesting prostacyclin, vasodilators or tadalafil [
46]. CD40L is an inflammatory factor that cleaves into sCD40L upon activation, which is known to promote significantly in PH patients, eventually contributing to vascular remodeling in PH [
47]. Although substantial evidence suggests that platelets and thrombogenicity exacerbate the pathogenesis of PAECs dysfunction, the molecular mechanisms need further elucidation.
5. PAECs Metabolism and Epigenetics in Intima Remodeling
Factors affecting PAECs metabolism and epigenetics participate in the regulation of PH. Metabolic abnormalities, particularly aerobic glycolysis or the Warburg effect, have been proposed as important pathogenic mechanisms in developing PH. PFKFB3 is an essential regulator of glycolysis, and its deficiency inhibits pulmonary vascular remodeling [
48]. Endothelin 1/eNOS signaling also serves as an essential pathway that regulates the glycolytic process [
49]. Further studies suggest that BolA family member 3 (BOLA3) is involved in the operation of glycolysis and mitochondrial respiratory function [
50]. Epigenetic mechanisms are also generally considered to be important in regulating PAECs metabolism. The delivery of glutamine carbon into the tricarboxylic acid (TCA) cycle becomes active in PAECs in the case of mutations in the BMPR2 gene, and the strict requirement for glutamine is driven by the loss of deacetylase sirtuin 3 activities. Additionally, the pharmacological effects of glutaminase can be targeted to reduce the severity of PH pathology [
51]. The distribution of other genetic variants showed that variants in ACVRL1, ENG, SMAD9, KCNK3 and TBX4 contributed to PH [
52], but only about 1% of the cases in each gene. Therefore, these factors are emerging as promising targets for PH treatment.
This entry is adapted from the peer-reviewed paper 10.3390/jpm13020366