Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Oncology
Toll-like receptors (TLRs) are indispensable for the activation, maintenance and halting of immune responses. TLRs can mediate inflammation by recognizing molecular patterns in microbes (pathogen-associated molecular patterns: PAMPs) and endogenous ligands (danger-associated molecular patterns: DAMPs) released by injured or dead cells. For this reason, TLR ligands have attracted much attention in recent years in many cancer vaccines, alone or in combination with immunotherapy, chemotherapy and radiotherapy (RT).
- cancer
- radiotherapy
- toll-like receptors
- innate immunity
1. Introduction
Toll-like receptors (TLRs) are central to the initiation of innate and adaptive immune responses. TLRs belong to the family of pattern recognition receptors (PRRs), which recognize molecular patterns in microbes (PAMPs) and endogenous ligands (DAMPs) [1]. There are 10 known TLRs in humans that recognize microbial ligands and products of damaged tissues [2]. Broadly, TLRs can be expressed on the cell surface or in the endosome. Cell surface TLRs recognize the lipopolysaccharide (LPS) of gram-negative bacteria (TLR4), bacterial lipoproteins (TLRs 1,2,6) and flagellin (TLR5) [3]. Endosomal TLRs mainly recognize nucleic acids, such as double-stranded RNA (TLR3), single-stranded RNA (TLR7) and double-stranded DNA (TLR9). With the exception of TLR3, TLRs activate the myeloid differentiation factor 88 (MyD88) signaling pathway. TLR3 signals in a TIR-domain-containing adapter-inducing interferon-b (TRIF)-dependent manner. TLR4 is the only receptor that can induce both MyD88 and TRIF-dependent signaling pathways. These pathways induce the synthesis or release of pro- and anti-inflammatory cytokines via the activation of transcription factors such as NF-κB, interferon regulatory factor (IRF)-3/7, AP-1 and others [4,5]. Such cytokines are often responsible for the recruitment of intratumoral immune cells and the resulting tumor immune phenotypes [6]. Given their role in mediating immune responses, the role of TLRs in tumor progression and tumor response to therapy remains poorly understood. In particular, the response to radiotherapy involves the induction of various types of cell damage and/or death which can activate TLRs and initiate downstream signaling. In this review, we cover the different roles of TLRs in cancer, and explore the mechanisms by which TLRs can lead to tumor progression or regression. Several prior review papers have focused on clinical aspects related to TLRs in cancer [7,8,9,10]. In addition, a recent clinical trials watch summarized the clinical studies to date that integrate TLRs in the treatment of cancer patients [11]. The reader is referred to those publications for clinical insight into TLR-based treatments. Here, we discuss the mechanistic effects of radiation on the expression of TLRs, and address how radiotherapy and TLR-based treatments can improve the radiotherapy response in solid tumors (Figure 1).
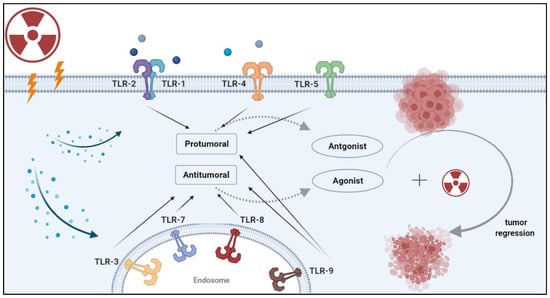
Figure 1. Strategies for combining radiotherapy with TLR-based immunotherapy. Radiation induces the release of damage-associated molecular patterns (DAMPs) by damaged or dying cells. DAMPs can be recognized by pattern recognition receptors (PRRs) such as TLRs. Once DAMPs bind to these receptors, they initiate a signaling cascade that leads to a pro-tumor or anti-tumor effect, depending on the specific TLR and the cancer type. Rationale combinations of TLR-based therapy with RT can enhance tumor recognition by antigen-presenting cells and lead to durable antitumor immunity. Image created with BioRender.com.
2. Activation and Inhibition of TLRs in Cancer
TLRs are expressed in a variety of cells, including T lymphocytes, monocytes, dendritic cells, alveolar epithelial cells, smooth muscle cells and fibroblasts [12]. Tumor cells have also been shown to express TLRs in ways that promote tumor growth, invasion, anti-apoptotic activity and treatment resistance [13,14]. At the time of this review, eight TLRs (TLR 1, 2, 3, 4, 5, 7, 8 and 9) have been found to be expressed in solid cancer cell lines, including lung, breast, head and neck, colorectal and pancreatic [15]. These are briefly reviewed below and summarized in Table 1.
Table 1. Summary of pro and anti-tumoral effects of TLR ligation in cancer. The number of studies for each cancer site is provided in brackets.
| Receptor | Ligand | Pro-Tumoral Effect (# of Studies) | Anti-Tumoral Effect (# of Studies) | Contributing Factors |
|---|---|---|---|---|
| TLR1/2 | BLP | Colorectal (2); Gastric (1); Ovarain (1) | Lung (3); Melanoma (1) | Effects are modulated through cancer-intrinsic mechanisms which vary depending on tumor type |
| TLR3 | Poly (I:C) | Esophageal (1); Breast (1); Gastric (1); HNSCC (1) | Lung (6); HNSCC (2); HCC (5); CRC (2); Neuroblastoma (1); Clear cell carcinoma (1); Breast (1); Cervical (1); Prostate (1); Melanoma (1) | Primarily anti-tumorigenic effects that are cancer-intrinsic and/or involve dendritic cells and CD8 T cells. Few correlative and in vitro studies suggest a pro-tumoral effect. |
| TLR4 | LPS | Lung (3); Breast (5); PDAC (1) | - | Pro-tumorigenic effect that is primarily cancer-cell intrinsic with limited immune involvement. |
| TLR5 | Flagellin | - | Gastric (1); CRC (1); Breast (3); Prostate (1) | Anti-tumorigenic effect mediated through neutrophils (likely N1). |
| TLR7/8 | ssRNA | PDAC (1) | Sarcoma (1); PDAC (1); Lung (1); CRC (1); Melanoma (1) | Anti-tumorigenic effect mediated through NK and CD8 T cells and decreasing MDSCs and Tregs |
| TLR9 | CpG | PDAC (1) | Melanoma (2); CRC (1); PDAC (2) | Mainly anti-tumorigenic effects mediated through recruitment of cDCs and priming of CTLs |
3. The Effects of Radiation on TLRs
The majority of TLRs are activated by microbial ligands. However, a wide variety of molecules of endogenous origin have also been reported to engage TLRs and activate them in the absence of microbial challenge. This includes small and large RNAs, DNA, ATP, UTP, chromatin, histones, mitochondrial DNA, heat shock proteins and calreticulin [80,81,82,83]. Despite the scarcity of basic studies investigating radiation and TLRs in cancer, radiation is recognized as either a direct stimulant of TLR pathways, or indirectly through the damage it causes to target cells that subsequently activate TLRs. Curtin JF et al. showed that 20 Gy irradiation of GL26 (glioma), LLc1 (lung carcinoma), GL261 (glioma) and B16-F10 (melanoma) cells increases HMGB1 expression [84]. The authors showed that HMGB1 is an endogenous agonist for TLR2, which is released from dying tumor cells in response to radiation or other stress-inducing agents (e.g., temozolomide). Treatment of syngeneic intracranial gliomas using a combination of FMS-like tyrosine kinase 3 ligand (Flt3L) and conditional cytotoxic gene thymidine kinase (TK) resulted in significant tumor regression. The effect was mediated through the MyD88/TLR2/NFκB pathway, and resulted in the infiltration of DCs into the brain parenchyma and the activation of cytotoxic CD8 T cells. By blocking HMGB1 activity, Flt3L/TK-induced brain tumor regression was inhibited. However, the authors did not perform similar experiments using RT alone or in combination with Flt3L or TK. Given their published data, it is reasonable to hypothesize that tumor-derived HMGB1 induced by RT elicits endogenous TLR2 signaling and initiate a CD8+ T cell-dependent immune response. In contrast to TLR2, TLR1 activation by RT has been shown to have tumor-promoting effects. Ryu et al. showed that macrophages exposed to irradiated tumor cells (CT26 cell line) had increased iNOS activity, increased nitric oxide (NO) production and increased arginase activity [85]. Knockdown of TLR1 decreased iNOS and NO activity, while TLR1 overexpression had the opposite effect. The authors fell short of testing the combination of TLR1 inhibition with RT.
In addition to HMGB1, an important mediator of TLR activity is p53. Shatz M et al. studied the dependency of TLR expression on the induction of p53 in response to radiation (among other stress-inducing agents). They showed that 10 Gy irradiation of cancer cell lines (U2OS, A549, HEPG2 and MCF7) increased the expression of TLR2, TLR3, TLR4, TLR6 and TLR9. The expression of TLRs 2, 3 and 9 was observed to be p53-dependent, and introduction of p53 into p53 null cancer cell lines induced expression of these TLRs [86]. This effect was not investigated by the authors in vivo, and it remains to be seen how TLR signaling and tumor p53 status influences the response to RT.
Another mediator of TLR signaling with relevance to radiation is IRAK2. IRAK2 is an immune-responsive protein kinase and a transducer of the IL1/TLR signaling cascade. In an analysis of radioresistant and radiosensitive isogenic oral squamous cell carcinoma cell lines, Yu CC et al. showed that IRAK2 expression is decreased in the radioresistant cell line at baseline and after radiation (4 Gy) [87]. In vivo, mice inoculated with the cell line overexpressing IRAK2 had significantly reduced tumor growth at baseline and in response to RT (50 Gy/10 fractions) compared to mice implanted with the parental radioresistant cell line (low IRAK2). The study also showed that patients with higher IRAK2 expression had a better recurrence-free survival rate. The effects of IRAK2 were shown to be mediated through apoptosis, as overexpression of IRAK2 increased apoptosis in response to radiation through cleavage of caspases 3 and 8 in the irradiated cells. Additional studies are warranted to investigate how IRAK2 is mediated by TLR signaling in response to radiation.
Based on the few studies to date on radiation and TLRs in cancer, it can be assumed that radiation-damaged cells release DAMPs which bind to TLRs and activate canonical inflammatory pathways. In particular, HMGB1 is a major component of dying cancer cells after radiation, and it can drive NF-κB and ROS production via activation of TLRs.
This entry is adapted from the peer-reviewed paper 10.3390/vaccines11040818
This entry is offline, you can click here to edit this entry!