Sanfilippo syndrome is caused by mutations in the enzymes responsible for the degradation of heparan sulfate (HS), a specific GAG, and patients are characterized by severe neurological pathology leading to childhood dementia.
- Sanfilippo syndrome
- mucopolysaccharidosis III
- lysosomal storage disorders
- heparan sulfate
- animal models
- induced pluripotent stem cells
- cellular models
- therapeutic approaches.
Sanfilippo syndrome
1. Introduction
Lysosomal storage disorders (LSDs) comprise a heterogeneous group of rare inherited metabolic diseases that are characterized by the accumulation of macromolecules inside lysosomes. LSDs are caused by deficiencies in lysosomal enzymes, leading to lysosomal dysfunction, altered recycling of macromolecules, and impaired flux of the endolysosomal system. Mucopolysaccharidoses (MPS) are a group of LSDs accounting for approximately 30% of all LSD cases and arise from mutations in genes involved in glycosaminoglycans (GAGs) degradation, which accumulate inside the lysosomes [1]. Among MPS, Sanfilippo syndrome (also known as mucopolysaccharidosis III or MPS III) is the most frequent type and it was first described more than 50 years ago [2].
2. Classification
There are four different subtypes of Sanfilippo syndrome based on the mutated gene and the consequent enzyme deficiency: type A (OMIM#252900), type B (OMIM#252920), type C (OMIM#252930), and type D (OMIM#252940), all of them presenting an autosomal recessive inheritance pattern [3]. Insufficient or complete loss of activity of any of the Sanfilippo syndrome causative enzymes leads to accumulation of partially degraded HS chains within lysosomes of cells in several organs and tissues [1,3,7]. In a recent study, a fifth subtype was identified in a mouse model [8] caused by mutations in the ARSG gene; however, to date, no human cases have been described. Moreover, human patients with a homozygous mutation in ARSG present Usher syndrome, leading to deaf-blindness and a small increase in urinary GAGs, although not as dramatic as in Sanfilippo syndrome patients [9].
Clinical symptomatology of Sanfilippo patients is similar regardless of the subtype, mainly characterized by an early-onset, severe, and progressive degeneration of the CNS with mild somatic symptoms [1,3,7]. Neurodegeneration starts during the first decade of life, with cortical atrophy, progressive dementia, motor deterioration, hyperactivity, learning difficulties, aggressive behavior, sleeping problems, and pronounced mental retardation [3]. Mild somatic manifestations include hirsutism, hepatosplenomegaly, joint stiffness, dysphagia, hypertrichosis, hypoacusia, speech loss, and skeletal alterations [1]. Death usually occurs at the second or third decade of life, although in unusual attenuated cases, life expectancy extends until the fifth or sixth decade [10–14].
The incidence of Sanfilippo syndrome varies depending on the subtype and geographical region, but on average is around one in 70,000 live births [15]. However, this incidence may underestimate the actual prevalence of different MPS III types because of the difficulties in the correct diagnosis of mild forms. Prevalence of the different subtypes vary between populations; subtype A being more frequent in the Northern Europe and subtype B more frequent in Southern Europe [16]. On the other hand, subtype C is in general less common while subtype D is very rare in all populations.
Table 1. Distribution of total mutations described for each Sanfilippo syndrome (MPS III) subtype (HGMDProfesional 2020.3; assessed on 9 October 2020).
|
|
Total Mutations |
Missense/Nonsense |
Small Deletions |
Small Insertions |
Small Indels |
Splicing |
Gross Deletions |
Gross Insertions and Duplications |
Complex Rearrangements |
|
A (SGSH) |
155 |
118 |
20 |
9 |
1 |
3 |
3 |
1 |
0 |
|
B (NAGLU) |
229 |
167 |
29 |
16 |
1 |
8 |
4 |
4 |
0 |
|
C (HGSNAT) |
77 |
43 |
6 |
6 |
1 |
15 |
4 |
1 |
1 |
|
D (GNS) |
25 |
7 |
5 |
4 |
1 |
4 |
2 |
0 |
2 |
2.1. Subtype A
MPS IIIA or Sanfilippo syndrome type A is caused by mutations in the SGSH gene, coding for sulfamidase (also known as heparan sulfate sulfatase or N-sulfoglucosamine sulfohydrolase, EC 3.10.1.1), which releases sulfate groups linked to the amino group of glucosamine. The gene is localized at 17q25.3 [17] with an approximated length of 11 Kb and contains eight exons. It codes for a protein of 502 amino acids with five possible glycosylation sites and a total of 155 identified mutations (Table 1). Sanfilippo syndrome type A is considered the most aggressive form, with patients surviving until 15–18 years old on average [16].
2.2. Subtype B
MPS IIIB or Sanfilippo syndrome type B is caused by mutations in the NAGLU gene, which encodes N-acetyl-α-glucosaminidase (EC 3.2.1.50), a lysosomal enzyme of 720 amino acids with six possible glycosylation sites. The function of the enzyme is the hydrolysis of the linkage between N-acetylglucosamine (GlcNAc) and the uronic acid, the two saccharides that conform HS. The gene maps to 17q21.2 [18]; spans 8.3 Kb; contains six exons; and, to date, 229 mutations have been identified as shown in Table 1. Sanfilippo syndrome type B patients die on average between 17–19 years old, this subtype being slightly less aggressive than subtype A [16].
2.3. Subtype C
Mutations in the HGSNAT gene are responsible for MPS IIIC or Sanfilippo syndrome type C. This gene codes for the lysosomal membrane protein known as acetyl-CoA α-glucosaminide N-acetyltransferase (EC 2. 3.1.78). It is located at chromosome 8p11.1, was identified by two independent groups in 2006 [19,20], spans about 62.5 Kb, containing 18 exons, and gives rise to a protein of 635 amino acids. For some time, there was controversy about the real initiation codon [21], but a recent publication suggested that only one ATG codon worked as the initiation codon [22]. Until now, 77 mutations have been identified (Table 1). Subtype C is the less aggressive form of Sanfilippo syndrome, with a mean survival of 19–34 years depending on the study [16].
2.4. Subtype D
Mutations in the GNS gene, which encodes the lysosomal enzyme N-acetylglucosamine-6-sulfatase (EC 3.1.6.14), are responsible for MPS IIID or Sanfilippo syndrome type D. The gene is located at 12q14.3, is 46 Kb-long, and contains 14 exons. The enzyme has 552 amino acids and 13 potential glycosylation sites [23]. It catalyzes the sulfate removal in the N-acetylglucosamine residues. Until now, 25 mutations have been found (Table 1). Due to the rarity of this subtype, there is no data on average survival of patients.
3. Therapeutic Approaches
Currently, there is no treatment to effectively slow down or reverse Sanfilippo syndrome patients’ neurodegeneration, and their management consists only of palliative measures to alleviate the symptomatology. Interestingly, different kinds of approaches have been tested during the last years in cellular and animal models of the disease, focused mainly on the treatment of the CNS involvement. The main approaches we will review here consist of enzyme replacement therapy (ERT), substrate reduction therapy (SRT), pharmacological chaperones, stem cell transplantation, and gene therapy (Figure 2). However, other approaches such as the use of coenzyme Q10 [66]; overexpression of TFEB [67], the master regulator in the lysosome biogenesis [68,69]; or the use of modified RNAs to recover aberrant splicing processes [70] have also been assayed showing different potentials to ameliorate pathological features of cellular and animal models.
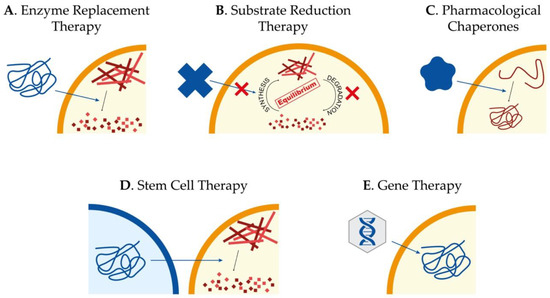
Figure 2. Potential therapeutic approaches to treat Sanfilippo syndrome. Schematic representation of the main therapeutic strategies currently being studied for the treatment of Sanfilippo syndrome patients: enzyme replacement therapy to provide the correct form of the mutated protein (A), substrate reduction therapy to reduce storage of undegraded molecules (B), use of pharmacological chaperones to correct protein missfolding (C), stem cell therapy for regeneration and production of the correct form of the protein (D) and gene therapy to provide cells with the correct form of the mutated gene (E).
3.1. Enzyme Replacement Therapy
The success of any therapy relying on administration or production of the correct form of the lysosomal enzyme relies on the fact that these proteins are tagged with mannose 6-phosphate (M6P) for correct trafficking towards the lysosome. Considering that cells have M6P receptors in the membrane, lysosomal enzymes can be endocytosed and arrive to the lysosome to perform their function [71]. For non-neurological LSDs, exogenous administration of the correct form of the enzyme mutated in patients, known as ERT (Figure 2A), has been proven to be the most successful strategy [72]. However, for diseases affecting the CNS, the existence of the blood–brain barrier (BBB), which limits the availability of the enzyme in the brain, has to be taken into account. In addition, antibodies targeting the enzyme can be observed in treated LSD-patients, clearly reducing the efficiency of the ERT [73]. Thus, intravenous administration is not as useful as for other LSDs without CNS pathology, for which ERT is currently approved and in use. On the other hand, direct brain administration for the treatment of neurological disorders seems more beneficial [74], although it is an aggressive treatment that needs continued injections. Nevertheless, clinical trials based on ERT for Sanfilippo syndrome type A and B have been carried out without clear results [75–78]. In any case, research to further investigate the potential of this approach is required [79].
3.2. Substrate Reduction Therapy
Taking into account the limitations of ERT, SRT has been presented as a valid alternative approach. The objective of this therapy is to find molecular targets to decrease the production of the accumulated substrate and restore the balance between synthesis and degradation (Figure 2B). It is important to remark that the mutant enzyme has to maintain some residual activity in order to achieve this restoration. SRT has been already approved to treat some LSDs, both with neurological and non-neurological symptomatology [80,81]. For Sanfilippo syndrome, different molecules with the ability to cross the BBB for the treatment of the CNS have been tested.
One of the most studied of these molecules is genistein, a natural isoflavone that inhibits the kinase activity of epidermal growth factor receptor, which is important for complete expression of genes encoding enzymes responsible for GAG production. Genistein was able to reduce GAG production in Sanfilippo syndrome type A and B fibroblasts [82], and to improve behavioral abnormalities, neuroinflammation, synaptic loss, and lysosomal storage in a Sanfilippo B mouse model [83]. After these positive results, two clinical trials with genistein treatment were carried out showing a reduction in urinary GAGs, but with unclear neurological benefits [84,85]. Another clinical trial using a higher dose of genistein was recently completed for Sanfilippo syndrome types A, B, and C. Even though these doses were safe for the patients, only a slight reduction of HS in the cerebrospinal fluid was observed, with no attenuation of the intellectual disability [79]. Further studies with higher doses of genistein and other flavonoids should be carried out to establish the ability of this group of molecules to ameliorate CNS pathology in Sanfilippo patients.
A different and interesting option for SRT is the use of specific RNAi directed to key genes involved in the GAG synthesis such as EXTL genes or genes involved in the linkage region formation. RNAi is a mechanism to selectively silence the expression of a particular gene by the specific degradation of the mRNA. Synthetic siRNAs and shRNAs have been widely used to downregulate the expression of a large number of genes in several cell types in vitro and in vivo. In one study, siRNAs were used to downregulate XYLT1, XYLT2, GALTI, and GALTII, genes encoding enzymes responsible for the formation of the linkage region [86] (Figure 1). This strategy was assessed in MPS I and MPS IIIA fibroblasts, resulting in an important decrease at the mRNA and protein levels for all the genes and a consequent significant decrease in the GAG synthesis after three days of treatment. In another study performed in our lab, the use of shRNAs to downregulate EXTL2 and EXTL3 genes was found to reduce the GAG synthesis and storage in MPS IIIA fibroblasts. These results were observed after three days of treatment, but failed after seven days [87]. Later on, fibroblasts from Sanfilippo C patients treated with similar siRNAs targeting EXTL2 and EXTL3 genes showed a reduction in GAG synthesis after three days and a decrease in HS storage after two weeks [88]. However, these studies were performed on patients’ fibroblasts, therefore, it is important to study SRT in relevant human neural cells, which are the ones affected in patients. In a recent study, we demonstrate that the same siRNAs that were effective in Sanfilippo syndrome type C fibroblasts were not efficient in decreasing storage in iPSC-derived neurons generated from the same fibroblasts assayed in the previous study [64].
3.3. Pharmacological Chaperones for Enzyme-Enhancement-Therapy
In many cases, missense mutations lead to the production of misfolded proteins that are rapidly degraded due to misfolding but that conserve some residual activity [89,90] and chaperons are cellular proteins that help proteins to adopt correct foldings. For years, several small compounds that act as chaperones, preventing misfolding of mutant proteins, have been identified (Figure 2C). Among the most common pharmacological chaperones that have been used for enzyme-enhancement therapy are amino and iminosugars. These molecules are in fact enzyme inhibitors that interact specifically with the active site of proteins and, used at low concentrations, can effectively stabilize the mutant enzymes and restore the correct folding to facilitate their trafficking towards the lysosome, thus, partially restoring enzymatic activity. In the case of LSDs, it has been proposed that achieving an enzyme activity around 5–15% can be sufficient to avoid the appearance of pathological symptoms [91]. To date, several compounds with chaperone activity have been tested for different LSDs such as Fabry disease, GM1-gangliosidosis, Morquio B disease, Pompe disease, Gaucher disease, Krabbe disease, Niemann-Pick A/B and C diseases, as well as for other types of disorders such as retinitis pigmentosa, cystic fibrosis, Parkinson’s disease, Alzheimer disease, or cancer [92].
In the case of Sanfilippo syndrome, several compounds were tested in a study for their potential to act as pharmacological chaperones [93]. The results showed that glucosamine, a competitive inhibitor of the HGSNAT enzyme with low toxicity, significantly increased HGSNAT activity in most patient fibroblasts lines tested, indicating its therapeutic potential. For Sanfilippo syndrome type C, we carried out a preclinical cell-based study showing a 2.5-fold increase of HGSNAT enzyme activity using glucosamine in patients’ fibroblasts carrying one splicing mutation that produces a protein lacking four amino acids [70]. Further studies should be done in order to establish its efficacy and lack of toxicity in brain cells as well as its ability to cross the BBB.
3.4. Stem Cell Therapy
In the last few years, several stem cell applications have been described for the treatment of neurological diseases in order to deliver the correct form of the enzyme into the brain (Figure 2D). Allogeneic bone marrow transplantation is used in the treatment of different LSDs with neurological pathology, but in the case of MPS III, intravenous administration of lentiviral-transduced bone marrow stem cells were not efficient to treat a mouse model of MPS IIIA [94] due to an insufficient production of enzyme by the donor cells or an inefficient uptake by the host cells [95].
Hematopoietic stem cell transplantation has been largely tested in many patients suffering from different LSDs. In patients’ brain, these cells can replace microglia and become enzyme-secreting donor cells [96]. Nevertheless, this process seems to be slow and not complete, making this option an invalid therapy for neurological disorders with a rapid progress of symptoms such as Sanfilippo syndrome, and currently, this approach is no longer considered for the treatment of Sanfilippo syndrome [97]. However, recent works using genetically modified hematopoietic stem cells carrying the normal copy of the SGSH or NAGLU genes showed an improvement in the neurological pathology in MPS IIIA or MPS IIIB mouse models [98–101].
Administration of human umbilical cord blood cells to the MPS IIIB mouse model has been explored, resulting in an amelioration of the neurological and somatic symptoms [102]. However, it presents the inconvenience that the enzyme production declines with time. On the contrary, the transplantation of umbilical cord blood-derived stem cells in two type B patients before the disease onset did not prevent the neurological deterioration [103].
Direct cell transplantation in the brain can be useful to both serve as cell replacement therapy addressing neuronal loss, as well as a source of cells secreting the correct version of the deficient enzyme [104]. In the last years, the development of iPSC technology has allowed researchers to easily generate patient-specific neural stem cells (NSCs), which have the potential to give rise to neurons, astrocytes, and oligodendrocytes. After transplantation into murine brains, NSCs can migrate long distances within the brain, differentiate, and integrate in the host network without disrupting normal functionality. In conclusion, NSCs represent an extraordinary opportunity to distribute the wild-type (WT) lysosomal enzyme and to recover neurological pathology, as it has been shown in studies in which MPS VII [105] and MPS IIIB [106] mouse models were treated with this strategy. However, for Sanfilippo C it is important to consider that HGSNAT does not have a M6P tag and is a membrane protein, therefore secretion and uptake of this enzyme by deficient cells may not be successful.
The use of glial precursors cells (GPCs) derived from pluripotent stem cells is another potential therapy to treat LSDs. In the mouse model of MPS IIIA [107], GPCs genetically modified to overexpress the SGSH gene were tested. Results showed promising results for this therapeutic approach, with GPCs successfully engrafting and surviving in the host brain, not forming teratomas, and showing long-term SGSH overexpression. Interestingly, astrocyte-based therapies are emerging as an option to treat some neurodegenerative disorders in which astrocytes play important roles, such as amyotrophic lateral sclerosis [108].
3.5. Gene Therapy
Gene therapy consists of the delivery of the correct copy of the gene to affected cells in order to recover enzyme activity (Figure 2E). Gene therapy is the most promising therapeutic option for LSDs since, as already referred, it has been proposed that only 5–15% of enzyme activity is required to maintain a healthy condition in affected patients [72]. Several clinical trials are currently ongoing or scheduled for different MPS [79]. In the case of Sanfilippo syndrome, several viral vectors have been tested for their therapeutic potential, such as retroviruses, lentiviruses, adenoviruses, and adeno-associated viruses (AAV). In addition, an approach using a nonviral vector (pFAR4) via tail vein administration was shown to increase enzyme activity and reduce GAGs storage in several tissues and lysosomes in the brain of an MPS IIIA mouse model [109]. Importantly, authors showed that liver of treated animals was converted into an enzyme distributor that promoted the GAG decrease in other tissues.
In the last years, the use of AAV have become the gold-standard tool for gene therapy in neurological disorders. Among the qualities that make them good vectors, it is important to highlight that they are nonintegrative, nonpathogenic, and nonimmunogenic in humans, and have the capacity to infect nondividing cells providing long-term expression. However, a recent study shows that AAV can induce cell death in some neural cell types in the murine hippocampus, suggesting that these approaches should be carefully evaluated [110]. Nevertheless, in the last few years, several reports have been published concerning AAV-mediated therapy using different virus serotypes and delivery strategies.
For MPS IIIA, intracerebral administration of AAV5 carrying the SGSH gene together with the SUMF1 gene (coding for an essential and limiting factor for sulfatases) in the mouse model showed an increase in the SGSH activity in the brain, a decrease in the storage and inflammation, and an improvement in the motor and cognitive function [111]. After these results, a phase I/II clinical trial for MPS IIIA using AAV10 expressing the deficient SGSH enzyme and the SUMF1 enzyme was started. It recently finished, showing no toxicity or lack of tolerance and a possible slight improvement in patient behavior [112]. AAV5 has also been used in another clinical trial with MPS IIIB patients, and results indicate an improvement of neurocognitive progression in all patients [113].
AAVrh10 has also been used to deliver SGSH in MPS IIIA mice via intraparenchymal administration [114]. This treatment reduced HS and GM3 ganglioside accumulation and microglial activation, but only in the site of injection. To increase efficacy, multiple intraparenchymal regions should be injected to ensure widespread distribution. To study SGSH distribution in the brain of large animals, the same transducing vector was injected via parenchyma in dogs and cynomolgus monkeys, and SGSH enzyme activity increase was detected [115].
In a study comparing delivery efficiency of the NAGLU gene using different AAV serotypes in MPS IIIB mice [116], a better biodistribution and transduction was found using AAV8 via direct administration of the virus to the CNS, but AAV9 showed better results for systemic or intracerebroventricular delivery. Intramuscular administration of AAV8 carrying the SGSH gene in Sanfilippo A mouse models showed no amelioration, while intravenous administration was effective in transducing mainly the liver, with a consequent amelioration of the pathology in somatic tissues, although with a discrete improvement in CNS symptoms of male mice [117]. To improve secretion and targeting of the CNS, another study used a fusion protein of SGSH with a signal peptide to boost enzyme secretion and a BBB-binding domain. This vector was administered with an AAV8, and results showed an important increase in enzyme activity in the brain that resulted in brain pathology and behavior improvements [118].
Recently, the safety of intravenous administration of an AAV9 carrying the NAGLU gene was tested in unaffected primates [119]. AAV9 has been suggested to be the most efficient serotype for targeting brain cells and therefore, for the treatment of neurological disorders. Very interestingly, a consistent and long-term increase in brain enzymatic activity was detected together with low immunogenic reaction. Similar successful results using AAV9 have been achieved in mouse and canine models of MPS IIIA [120,121]. First, a clear increase in enzyme activity combined with a reduction in GAG storage and neuroinflammation was found in the mouse model treated intravenously, resulting in expanded lifespan [121]. Later, both animal models were treated with intracerebrospinal injections, showing low immunogenic reaction and resulting in a clear restoration of enzymatic activity and full body reduction of GAG storage and lysosome alterations, leading to prolonged lifespan [120]. This same research group also develop a strategy to treat MPS IIIB [122] or MPS IIID [54] mice with cerebrospinal fluid delivery of AAV9 vector carrying NAGLU or GNS genes, respectively. After treatment, enzyme activity in the CNS, normalization of GAG storage, corrected behavior, and extended lifespan were observed.
All these results in different Sanfilippo subtypes encouraged the application of this approach in human patients. In relation to cerebrospinal fluid administration, Esteve Laboratories recently started a phase I/II clinical trial using AAV9-hSGSH in MPS IIIA patients (EudraCT Number: 2015-000359-26). Besides, although some preclinical studies have been performed before, it was recently confirmed that some AAV were able to cross the BBB [123]. Due to that, Abeona Therapeutics has started a clinical trial using an intravenous delivery of AAV9 vector carrying the human SGSH gene under the control of a U1a promoter (ClinicalTrials.gov: NCT02716246, NCT04088734). Preliminary data showed a dose-dependent and sustained reduction in cerebrospinal HS after 30 days. In the case of Sanfilippo syndrome types A and B, two clinical trials based on intracerebral injection of AAV have been already completed [112,113], and another two for subtype A have started (ClinicalTrials.gov: NCT03612869, EudraCT Number: 2015-000359-26). However, as for ERT, gene therapy success for lysosomal enzymes relies in the ability of transduced cells to share the correct lysosomal enzyme through M6P receptors with non-transduced neighboring cells [68]. As mentioned above, HGSNAT is a lysosomal transmembrane protein that does not undergo the M6P pathway. For this reason, Sanfilippo C syndrome might not be the best candidate for gene therapy strategy, although some interesting results have been obtained with a novel AAV [124].
This entry is adapted from the peer-reviewed paper 10.3390/ijms21217819