DiGeorge syndrome (DGS) is a rare genetic disease caused by microdeletions of the 22q11.2 region (DGS1). A haploinsufficiency at 10p level has been proposed also as a DGS cause (DGS2). Clinical manifestations are variable. The most frequent features are thymic hypoplasia or aplasia with consequent immune deficiency, cardiac malformations, hypoparathyroidism, facial and palatine abnormalities, variable degrees of cognitive impairment and psychiatric disorders. The specific aim of this descriptive report is to discuss the correlation between oxidative stress and neuroinflammation in DGS patients with microdeletions of the 22q11.2 region. The deleted chromosomic region maps various genes involved in mitochondrial metabolisms, such as DGCR8 and TXNRD2, that could lead to reactive oxygen species (ROS) increased production and antioxidant depletion.
1. Introduction
DiGeorge syndrome (DGS) or del22q11.2 syndrome is a genetic disease that involves microdeletions of the 22q11.2 (DGS1) [
1] and/or 10p13/10p14 boundary (DGS2) [
2,
3]. Along with the well-defined clinical manifestations, this syndrome might involve changes in the regulatory mechanisms of oxidative stress and neuroinflammation as shown in animal models [
4].
2. DiGeorge Syndrome
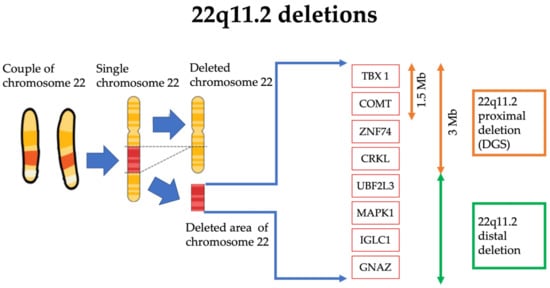
DGS is a genetic syndrome caused by a microdeletion of the 22q11.2 region. The incidence is about 1 in every 4000/6000 live births, although the real prevalence could be higher due to the lower diagnosis rate in developing countries [
5] (
Figure 1).
Figure 1. Schematic representation of the 22q11.2 deleted region of the chromosome 22 in DGS. The dimension of the deleted section (in red) is amplified for visual and graphical reasons.
The deleted 22q11.2 region encodes for 90 different genes, even if only some of the deleted genes appear to be directly related to the manifestations of the syndrome. In fact, one of the genes mostly correlated with the typical anomalies of the syndrome is TBX1, belonging to the transcription factors of the T-box family [
6]. This gene plays a key role in the genesis of mesoderm, endoderm and pharyngeal ectoderm. TBX1 loss involves anomalies in the correct genesis of the thymus, pharynx and mandibular region [
6]. Furthermore, TBX1 is involved in the development of the right ventricle and the arterial cone, which could explain the cardiological features frequently found in DGS. Along with heart abnormalities, TBX is also involved in cerebral microvascular system formation, which could explain the DGS patient susceptibility to developing psychiatric pathologies [
6,
7].
Another typically deleted gene is DGCR8, encoding the DGCR8 microprocessor subunit, a double-stranded RNA protein that mediates the synthesis of several miRNAs [
8]. Lacking DGCR8 alters the expression of genes adjacent to those of the deleted region, favoring the psychiatric manifestations associated with the syndrome [
9] as also shown in animal models [
10].
The DGS signs and symptoms are so varied that different combinations of its descriptions were once considered as independent conditions. These early classifications include also Shprintzen syndrome, DiGeorge sequence/syndrome, velocardiofacial syndrome, conotruncal anomaly face syndrome and Sedlackova syndrome [
9,
11,
12,
13,
14]. Sometimes immunodeficiency or immune manifestations are not pronounced, which is generally required for the diagnosis of DiGeorge syndrome, even in patients where velocardiofacial symptoms are present [
5,
15,
16]. All are now considered to be presentations of a single syndrome.
Indeed, the DGS clinical presentation is heterogeneous [
1,
7,
17,
18,
19,
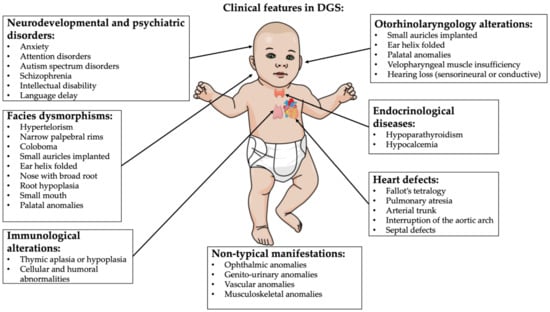
20]. The decline in quality of life is strongly related to congenital cardiac defects, psychiatric disorders and palatal anomalies. Recurrent infections and autoimmune disorders (thrombocytopenia being the most common manifestation) due to immunodeficiency and immune-dysregulation caused by thymic dysfunction and hypoparathyroidism/neonatal hypocalcemia related to parathyroid abnormal development consistently contribute to the clinical phenotype [
1]. Renal, vertebral and airway structural anomalies, hypotonia, growth and neurodevelopmental delay, microcephaly and hearing alterations are also present [
5] (
Figure 2).
Figure 2. DGS typical clinical features. Image created by mindthegraph.com platform.
Important features can be recapitulated using the mnemonic
CATCH-22 to indicate 22q11.2DS, with the 22 suggesting that the chromosomal aberration is located on the 22nd chromosome [
21]:
-
Cardiac abnormality (commonly interrupted aortic arch, truncus arteriosus and tetralogy of Fallot)
-
Abnormal facies
-
Thymic aplasia or hypoplasia
-
Cleft palate
-
Hypocalcemia/hypoparathyroidism early in life
DGS individuals may show many possible outcomes, fluctuating in number of associated outcomes and from the mild to the very severe. Symptoms demonstrated to be common comprise the following [
1,
7,
17,
18,
19,
20]:
- -
-
Palatal abnormalities (50%), particularly velopharyngeal incompetence, submucosal cleft palate and cleft palate; characteristic facial features (present in the majority of Caucasian individuals) including hypertelorism;
- -
-
Cyanosis (bluish skin due to poor circulation of oxygen-rich blood);
- -
-
Congenital heart disease (40% of individuals), particularly conotruncal malformations (interrupted aortic arch (50%), pulmonary atresia, persistent truncus arteriosus (34%), tetralogy of Fallot and ventricular septal defect);
- -
-
Hearing loss (both conductive and sensorineural) (hearing loss with craniofacial syndromes);
- -
-
Hypocalcemia (50%) (due to hypoparathyroidism);
- -
-
Significant feeding problems (30%);
- -
-
Learning difficulties (90%), including cognitive deficits and attention deficit disorders;
- -
-
Laryngo-tracheo-esophageal anomalies;
- -
-
Growth hormone deficiency;
- -
-
Renal anomalies (37%);
- -
-
Psychiatric disorders;
- -
-
Autoimmune disorders;
- -
-
Immunodeficiency present in about 75% of patients, related to thymic aplasia or hypoplasia determining alterations in both humoral and cell-mediated immune responses;
- -
-
Immune disorders due to reduced T cell numbers;
- -
-
Schizophrenia develops in 25–30% by adulthood;
- -
-
Seizures (with or without hypocalcemia);
- -
-
Skeletal abnormalities.
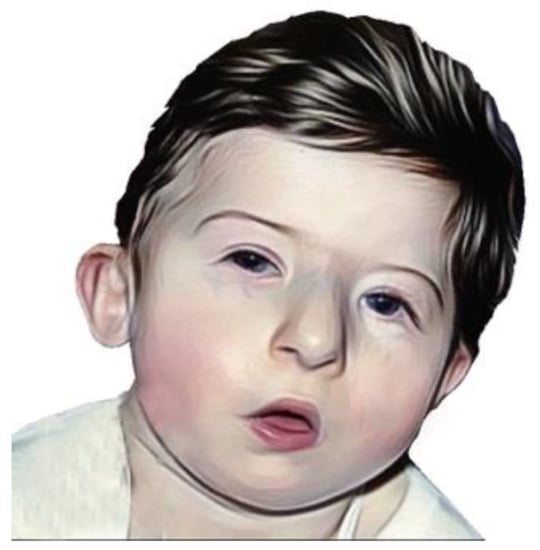
Facial features of DGS are quite typical and are represented by hypertelorism, narrow palpebral rims, coloboma, small auricles implanted down and rotated anteriorly, the helix folded, the nose with broad root and root hypoplasia, smallmouth and micrognathia which usually decreases with age [
22] (
Figure 3).
Figure 3. A DSG patient showing characteristic facial appearance, with tubular nose and carp-shaped mouth.
Psychiatric disorders may have a very negative impact on the quality of life of these patients and include anxiety, attention disorders, autism spectrum disorders and schizophrenia; the latter seems to affect between 25% and 40% of subjects with DGS [
23].
In addition, 20% of patients affected have unilateral facial paralysis due to hypoplasia of the depressor muscle in the corner of the mouth (DAOM) that causes an asymmetry of the lower lip, evident especially during crying [
5].
The diagnosis is genetic by performing array-CGH. This can be effectuated during pregnancy or after birth in case of clinical suspicion [
24]. Life expectation of these patients is reduced due to psychiatric and cardiological comorbidities related to the syndrome [
7,
23].
3. Oxidative Stress in DiGeorge Syndrome
Oxidative stress plays an important role in the development of different signs and symptoms of various genetic syndromes [
25]. The correlation between the imbalance of the redox state and the clinical DGS manifestations has not yet been established, but important findings have been revealed, as shown in
Table 1 [
26]. In particular, in 2019, Fernandez et al. used a mouse model of DiGeorge/22q11 deletion syndrome to describe biological mechanisms of neuronal under-connectivity, which reflects reduced growth of dendrite, axon and synapse [
4]. It seems that connections between and within regions of the cerebral cortex are the basis for complex behaviors [
27]. It has been proposed that the under-connectivity of association cortices underlies behavioral deficits in several neurodevelopmental disorders, including DGS [
28].
Table 1. Relations between genetic or enzymatic alterations and markers of oxidative stress in patients affected by DGS.
Observing functional images, it has been demonstrated that association cortices are under-connected in the DGS [
32]. Fernandez et al. demonstrated that under-connectivity occurs due to inefficient reactive oxygen species (ROS) catabolism and mitochondria-associated oxidative stress in layer 2/3 projection neurons (responsible for association cortico-cortical connections), while layer 5/6 neurons were unaffected [
4].
In addition, the reduced dosage of the gene Txnrd2, a 22q11 gene essential for reactive oxygen species clearance in brain mitochondria, is responsible for the disruption of layer 2/3 projection neurons’ antioxidant defense and cellular consequences.
They also demonstrated that the use of antioxidants or the re-expression of Txnrd2 improved quantitative cellular deficits associated with LgDel layer 2/3 PN-selective under-connectivity. Accordingly, the oxidative stress could be considered a therapeutic target in neurodevelopmental disorders, as in DGS, due to the connection between the antioxidant restoration of mitochondrial integrity, cortical connectivity and cognitive behavior [
4].
The relationship between DGS and alterations in the function of mitochondria has been also studied by Napoli et al. in 2015 [
8]. Under high-oxidative-stress conditions, the mtDNA copy number per cell and mtDNA deletions are usually increased. Napoli et al. found that the mtDNA copy number and mtDNA deletions were both increased in the peripheral blood monocytic cells in DGS patients. Thus, several plasma metabolites were also indicative of increased oxidative stress. Especially, they observed an increased flux through the pentose phosphate shunt and products of oxidatively modified proteins (such as Met sulfoxide from Met, indole-3-lactate from Trp and aminomalonate) and hexoses (5-hydroxymethyl-2-furoic acid, hexuronic acid) [
8].
In addition, they found that the elevation of 2HG in plasma could reflect a significantly higher intracellular (in particular intramitochondrial) concentration of 2HG. Accumulation of this metabolite has the potential to inhibit Complex IV, Complex V and creatine kinase and also increase oxidative stress, leading to a secondary block of the tricarboxylic acid (TCA) cycle.
This could result in a higher ratio of [NADH]/[NAD] and [FADH2]/[FAD], followed by the production of NADPH through the mitochondrial nicotinamide nucleotide transhydrogenase to sustain isocitrate dehydrogenase 2 activity and the reductive carboxylation of glutamine to citrate [
8]. Furthermore, they pointed out that 9 of the 30 genes involved in DGS have the potential of disrupting mitochondrial metabolism. They especially focused on the TXNRD2 gene.
TXRND2 is one of the six 22q11 mitochondrial genes, with PRODH and ZDHHC8, that have SNPs associated with schizophrenia [
31,
33]. TXNRD2 is elevated in schizophrenic-brain samples [
34]. The TXNRD2 is a gene that encodes for a mitochondrial thioredoxin reductase. This thioredoxin reductase catalyzes the reduction of the active disulfide of thioredoxin 2 and other substrates, having an important role in antioxidant defense [
35].
An altered dosage of one of the six 22q11 mitochondrial genes could influence mitochondria function causing changes in synaptic development or function that could provide to the increased susceptibility for psychopathology in DGS as schizophrenia [
36,
37]. Recently, the relationship between premature cellular senescence and mitochondrial oxidative stress has been investigated by silencing the gene of the critical region 8 of the DiGeorge syndrome (DGCR8), an essential component of miRNA biogenesis [
26]. Indeed, the authors studied the role of microRNAs (miRNAs) in regulating cellular senescence in human mesenchymal stem cells, depleting miRNAs in the DGCR8. They found that the human mesenchymal stem cells with the DGCR8 knockdown showed critical proliferation defects and alterations associated with senescence, including increased levels of ROS. Transcriptomic analysis studies revealed that the antioxidant gene superoxide dismutase 2 (SOD2) was significantly downregulated in DGCR8 knockdown hMSCs. They also discovered that DGCR8 silencing in hMSCs caused hypermethylation in CpG islands upstream of SOD2. The treatment with 5-aza-2′-deoxycytidine reestablished SOD2 expression and ROS levels [
26].
In conclusion, it is still not possible to describe a direct connection between oxidative stress and all the clinical manifestations of patients with 22q11DS deletion, but recent studies have pointed out that there could be a link between the psychiatric manifestations and alteration of crucial genes for the mitochondrial metabolism, leading to a reduction of antioxidant defenses. Further studies are needed to evaluate this connection and eventually the benefit of an antioxidant therapy.
This entry is adapted from the peer-reviewed paper 10.3390/ijms24044242