The discovery of cell-free deoxynucleic acids (cfDNA) in the sera of cancer patients in 1948, is attributed to Mandel and Métais [
1]. Later, a correlation was observed between the concentration of cfDNA and the development of systemic lupus erythematosus [
2]. The use of cfDNA in the diagnosis of tumors began in 1977, but was not very effective, due to the limitations of the existing technology [
3]. The real-time polymerase chain reaction allowed the detection of RhD and the fetal sex in maternal plasma in 1997 [
4]. The real expansion of non-invasive fetal genetic disease detection began in 2011, with the introduction of massive parallel sequencing [
5]. Approximately fifty percent of prenatal genetic examinations are performed today via so-called non-invasive prenatal testing (NIPT) [
6]. Recently, the spread of liquid biopsies and the increased demand for screening, as well as monitoring disease activity and the therapeutic response, made it possible for cfDNA research to be revived. Though the analysis of the 5′ ends of extracellular DNA demonstrated the unique character of extracellular DNA (i.e., definitely not being a junk molecule) [
7], investigations primarily focus on the role of cfDNA as a biomarker, and research regarding the immunological properties of cfDNA, such as its potential immunomodulatory or therapeutic benefits, is still in its infancy.
2. Origin, Release, Amount, and Clearance of Cell-Free DNA
Though cfDNA is ubiquitously present in human body fluids [
8], and many aspects of its molecular source are known, research to uncover the unknown factors in its origin is growing and may never end. Except for the exogenous sources of cfDNA [
9], many possible endogenous origins and related mechanisms have been proposed [
10]. Regarding the cellular source of cfDNA, tumorous (i.e., local and circulating tumor cells, micrometastases, and cells of the tumor microenvironment) and non-tumorous cells (e.g., muscle cells, epithelial cells, ovum cells, bone cells, myeloid and lymphoid cells) can be distinguished [
10].
The mechanisms responsible for cfDNA release are quite diverse. On the one hand, cell death and clearance mechanisms (i.e., apoptosis, necrosis, pyroptosis, mitotic catastrophe, autophagy, phagocytosis, oncosis, NETosis, and DNA excision repair damage) are partly responsible for the release of cfDNA [
11,
12]. On the other hand, the active release is also possible via macromolecular structures (DNA-protein complexes, extracellular traps), micronucleation induced by genome instability (extrachromosomal circular DNA), or microvesicles (exosomes) [
13,
14,
15].
Different data is available on the amount of human cfDNA in circulation since no standardized methods exist. The choice of matrix (i.e., serum, plasma, urine, cerebral fluid, etc.), the mode of sample collection (e.g., EDTA-containing tubes or CellSave tubes, etc.), the parameters of centrifugation (i.e., speed, temperature, duration), types of isolation kits, and cfDNA storage conditions can all influence the measurement results [
16]. In general, the level of cfDNA in the healthy population is lower, as compared with diseased people. According to the latest data [
17], the normal human plasma cfDNA concentration can be as high as 500 ng/μL. In cases of advanced cancers [
18,
19,
20], autoimmune [
21,
22,
23,
24,
25], inflammatory [
26], traumatic [
27,
28], post-transplantation [
29] or infectious diseases [
30,
31] usually a more increased amount is detected. In addition, cfDNA levels could also be increased, due to vigorous physical exertion (such as intense sports, e.g., half marathon, ultramarathon, TRX exercises) [
32,
33] and pregnancy [
34]. Fetal cfDNA, which is primarily produced by placental trophoblast cells during pregnancy [
35], is detected in the maternal circulation, as early as in the first trimester, accounting for 10 to 15% of the total cfDNA concentration [
36].
The concentration of cfDNA can increase, not only under the previously mentioned conditions, but also as a result of an increase in release. Ineffective clearance mechanisms could also contribute significantly to the elevated levels of circulating cfDNA. Extracellular nuclease homologs, DNase I and DNase I-like III (DNase I L3), are responsible for the efficient degradation of both free and protein-bound DNA [
37]. The enzyme’s ability to recognize and degrade DNA could be influenced by the abnormalities of DNase I activity (e.g., low serum DNase I activity [
38], elevated serum levels of DNase I inhibitors [
39], novel mutations in the enzyme [
40]), molecules that interact with DNA [
41], anti-DNase antibodies [
42,
43], and deficiencies in DNase I activating cofactors, such as the complement component C1q [
44], TREX1 DNase [
45], serum amyloid P component [
46], IgM [
47], C-reactive protein [
48], and mannan-binding lectin [
49]).
3. Cell-Free DNA as a Molecular Marker or a Diagnostic Tool
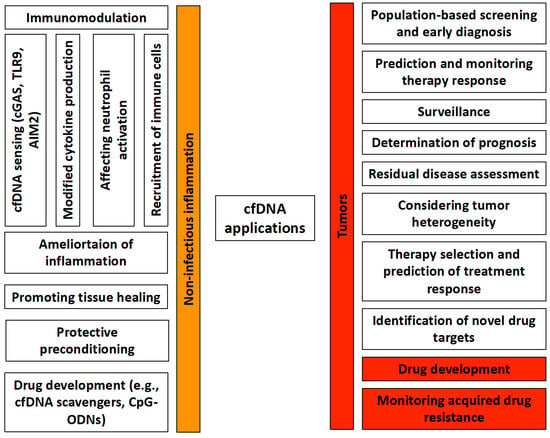
Based on its close association with a number of human physiological and pathological conditions, the clinical utility of cfDNA as a noninvasive, reliable, sensitive, and rapid diagnostic marker is continuously the subject of intense research (Figure 1).
Figure 1. Potential clinical and experimental applications of cfDNA in non-infectious inflammations and tumors. cfDNA has an immunomodulatory effect in non-infectious inflammations, which is mediated by cfDNA sensing, changes in cytokine production, neutrophil activation, and effects on other immune cells. cfDNA by itself can reduce inflammation, promote tissue healing, and is also suitable for protective pretreatments. In addition, it can serve as a promising starting point for drug development. In tumors, it can play a significant role in population-level screenings, early diagnosis, and therapeutic response determination. In addition to being able to predict the course of the disease, it can also be used to determine any residual disease after treatment. It provides information on the heterogeneity of tumor cells, thereby facilitating the selection of the most effective treatment. It can serve as a basis for drug development. Furthermore, it allows for the monitoring of acquired drug resistance.
3.1. cfDNA in Prenatal Diagnosis
Prenatal genetic testing is among the fields in which the utilization of circulating cfDNA has had the most success and is still widely used [
50]. NIPT became a clinical reality in 2011 [
51]. The fetal-derived cfDNA can be detected as early as the 4th week of gestation [
52], and it is quickly eliminated from the maternal bloodstream after delivery [
53], emphasizing its pregnancy specificity. The first clinical applications were limited to the identification of alleles present in the fetus and not in the maternal genome (i.e., paternal or de novo mutations) [
54]. In contrast, the establishment of autosomal recessive or maternally transmitted autosomal dominant disorders, has been much more complicated, even though several studies have succeeded in determining the exclusion of paternal alleles in recessive conditions. However, thanks to the continuous development of technology, it is now possible to determine the sex, RhD, and blood group of the fetus [
55]. In addition, cfDNA also allows the identification of fetal aneuploidies and specific microdeletions [
56]. Though the measurement of fetal cfDNA is noninvasive, widely applicable, and available early in pregnancy, it has some limitations. The current detection of aneuploidy is limited to common trisomies [
57], therefore the karyotype determination is still necessary. Furthermore, fetal cfDNA determination is currently irrelevant for diagnosing monogenic disorders, autosomal recessive, or X-linked diseases [
58]. So, the technique needs to be improved.
3.2. cfDNA in Tumors
The growing interest in tumor-related cfDNA is a direct result of its potential use as a liquid biopsy tool, which has great promise for a wide range of clinical applications [
59,
60]. Even if a surgical biopsy/histology remains the gold standard for cancer diagnosis and treatment, it has some disadvantages (i.e., it is invasive and provides temporary static images of malignancy) [
61]. In contrast, tumor cfDNA detection enables the real-time longitudinal monitoring of cancer, along with capturing tumor heterogeneity [
62,
63,
64]. Moreover, in the last few years, there has been a strong concordance between plasma and tissue-based genomic studies, encouraging the exploration of their potential clinical utility [
65,
66,
67,
68]. Tumor cfDNA has received a lot of attention in early tumor detection for several types of cancer [
69], however, the process for purification and handling of cfDNA is not yet standardized, and numerous preanalytical variables, such as the purification kits, blood collection tubes, and centrifugation regime, may affect cfDNA’s yield and analysis [
70,
71]. Thus, more sensitive and reproducible techniques are required. For screening, the combined use of tumor cfDNA and conventional tumor markers seems to be an optimal application [
72,
73,
74]. Several studies have demonstrated that in several types of cancer tumors, cfDNA is suitable for detecting minimal residual disease postoperatively or after chemotherapy [
75,
76,
77], which suggests that it has a high prognostic value with the ability to predict the disease recurrence. The genotyping of tumor cfDNA is useful, not only in choosing the optimal treatment and dynamically monitoring the therapeutic responses [
78], but it can also reveal the genetic causes of malignancy progression and therapy resistance, as well [
78]. Applications of tumor cfDNA in this direction seem feasible and are close to being introduced into clinical practice.
3.3. cfDNA in Non-Tumor Disorders
In pathological conditions, such as autoimmune diseases [
79,
80,
81,
82], stroke [
83], myocardial infarction [
84,
85], and allograft transplant rejection [
86], there is substantial interest in the investigation of cfDNA’s clinical utility, but no real medical applications have been developed yet. Elevated levels of cfDNA in SLE patients appear to be associated with antibody titers and active lupus nephritis [
82,
87], but its correlation with disease activity, as well as the diagnostic and prognostic values, remains uncertain [
83,
87]. In rheumatoid arthritis (RA) patients, the serum level of cfDNA seems to be quite varied [
81,
88,
89]. The cfDNA concentration of synovial fluid is several times higher than that in circulation, indicating the importance of local inflammation in the cfDNA release [
90]. In RA patients, the dynamics of cfDNA appear to be independent of the conventional diagnostic markers, ACPA and RF. Though studies suggest the biomarker potential of cfDNA, further studies with large patient cohorts are necessary to analyze the dynamics of cfDNA in RA, in relation to the disease progression and drug effects.
In cases of stroke, the dynamically determined blood levels of cfDNA appear to be a valid and reliable option for establishing prognostic and diagnostic criteria [
91]. While cfDNA has performed well in a number of studies as a stroke biomarker [
92,
93], none of the so-called stroke biomarkers identified to date have proven useful in medical practice, and there is still a long way to go before its clinical application, either as a standalone marker or as part of a biomarker panel.
cfDNA testing provides an alternative method for monitoring myocardial ischemia and has potential clinical applications for identifying high-risk individuals [
94]. However, several biological and technical obstacles were recognized in cell-free DNA testing [
95], including the lack of specificity and unsuitable kinetics for early cardiomyocyte damage, the long turnaround time and limited bandwidth, the need for specialized equipment and specialized staff, the absence of standardized or harmonized analytical techniques, the indirect expenses, and the high susceptibility to preanalytical variables [
95]. Therefore, it seems acceptable to conclude that the analysis of cell-free DNA in diagnosing myocardial ischemia is not yet ready for commercialization.
In organ transplantation, the diagnostic role of cfDNA has been extensively studied in heart, kidney, and lung transplantations [
96]. However, only one study exists on this topic in liver transplantation [
97]. Despite the many results supporting the association between the amount and kinetics of donor-derived cfDNA and transplant organ rejection, neither the US Food and Drug Administration nor the European Medicines Agency has approved the use of cfDNA in this context. Based on the objections, clarification is needed on both the threshold and kinetics.
4. Recognition and Immunomodulatory Role of Cell-Free DNA
In addition to being a biomarker and a diagnostic tool, cfDNA has been shown experimentally to have an immunomodulatory effect. It can influence the initiation, progression, or amelioration of inflammation. The presence of self-DNA in the nucleus and mitochondria is necessary for the maintenance of self-tolerance. However, following nuclear or mitochondrial damage, self-DNA enters the cytosol under stress conditions. In the apparent lack of infection, the inflammatory response is likely triggered by the production of endogenous alarmins, known as danger-associated molecular patterns (DAMPs), which trigger immune responses via pattern-recognition receptors (PRR). Cell-free DNA could act as a DAMP [
98,
99].
The recognition of cfDNA could be performed by the DNA-sensing receptor cyclic guanosine monophosphate (GMP)-adenosine monophosphate (AMP) synthase (cGAS), Toll-like receptor 9 (TLR9), or absent in melanoma-2 (AIM2)-like receptors (ALRs) [
100].
The cGAS identifies cytosolic DNA and induces the interferon regulatory factor (IRF) 3-dependent interferon-beta (IFNβ) or type 1 interferons [
101]. cGAS recognizes extracellular nucleosomes as well, because they have a higher binding capacity than double-stranded DNA (dsDNA) [
101]. Stimulator of interferon genes (STING) participates in the cGAS signaling pathway in response to the recognition of cytosolic DNA [
102,
103]. The cGAS optimally recognizes 36 base pair long dsDNA (or longer) to activate the cGAS-STING-mediated effectors to generate type 1 interferons and other nuclear factor (NF)-kB-dependent cytokines, regardless of the sequence [
100,
104,
105]. In addition to NF-kB, mitogen-activated protein kinase (MAPK), and signal transducer and activator of transcription 6 (STAT6) activation, STING stimulates the autophagosome formation by facilitating the microtubule-associated protein 1A/1B-light chain 3 (LC3) puncta formation and the autophagy related (Atg) 9a, upon recognizing the cytosolic dsDNA [
106,
107,
108,
109]. Beclin-1 (BECN1) interacts with cGAS to restrict cGAMP formation in response to cytosolic dsDNA, by inhibiting the interaction between cGAS and dsDNA. The interplay between cGAS and BECN1 results in the release of Rubicon (negative regulator of autophagy) from BECN1, which activates the class III phosphatidylinositol 3-kinase function to induce autophagy and hence eliminate cytosolic dsDNA [
110]. One of the main functions of autophagy is to eliminate cfDNA without causing inflammatory damage. The defected autophagy enhances the inflammatory recognition of cfDNA by various cytosolic PRRs [
110].
TLR9 is present in the endoplasmic reticulum (ER), during normal physiologic stages. However, when cytosolic cytosine-phosphate-guanine (CpG)-DNAs or self-DNA enter the endosome or endolysosome, TLR9 migrates to these organelles and recognizes them as essential DAMPs [
111,
112]. In order to cause inflammation and inflammatory diseases, the TLR9 activation induces a myeloid differentiation primary response 88 (MyD88)-dependent downstream signaling pathway that activates the IRF3-based type1-interferon production and NF-kB-mediated pro-inflammatory cytokine production [
113]. The Toll-interleukin-1 receptor (TIR) domain of MyD88 activates the interleukin 1 receptor-associated kinase (IRAK)-4 and IRAK-1 [
114,
115]. IRAK-4 recruits the tumor necrosis factor receptor-associated factor 6 (TRAF6) to activate the transforming growth factor-β-activated kinase 1 (TAK1) [
116]. TAK1 phosphorylates the IκB kinase (IKK) complex via the K63-linked ubiquitination of the NF-kB essential modulator (NEMO), which is crucial for the NF-kB, IRF3, and MAPK signaling [
117]. TLR9 recognizes two types of DNA (i.e., pathogen-derived and self-DNA). It was shown that the nucleotide sequence, length, and dimerization properties of synthetic CpG-oligodeoxyribonucleotides (ODNs) and cfDNAs determine their tendency to bind and activate TLR9 [
118,
119,
120]. The intracellular compartmentalization of TLR9 is a mechanism for discriminating between self- and non-self-DNAs [
121]. Their binding results in an increase in the dimerization and activation [
121].
Platelets are known to express PRRs, which can be triggered upon interaction with DAMPs [
122]. Platelets from both murine and human hosts express TLR9 [
123,
124], which is of importance because, in addition to their hemostatic function, platelets play a crucial role in bridging innate and adaptive immunological responses [
122]. Platelet activation results in the platelet production of P-selectin, which enables platelets to attach to other cells, such as granulocytes, leading to the granulocyte activation and recruitment to sites of tissue damage. Platelets are activated by cfDNA, which contributes to the creation of neutrophil extracellular traps (NETs) [
122].
AIM2 is an ALR that is activated upon recognizing and binding to self-DNA entering the cytosol, as a result of cellular damage and exosomes containing self-DNA [
125]. AIM2 efficiently activates in response to 80–300 base pair self-DNA [
126,
127]. The HIN (hematopoietic expression, interferon-inducible nature, and nuclear localization) domain of AIM2 recognizes cytosolic DNA, and its pyrin domain (PYD) interacts with the PYD of ASC (apoptosis-associated speck-like protein containing a C-terminal caspase recruitment domain) to form an inflammasome complex that converts procaspase 1 (pro-CASP1) to CASP1 [
126,
128]. CASP1 releases IL-1 and IL-18 from their preforms [
128]. CASP1 also cleaves the Gasdermin D (GSDMD) linker region, finally mediating the release of IL-1 and IL-18 from cells. Additionally, the K
+ efflux from the GSDMD pore inhibits the cGAS activity and the cGAS-STING-mediated release of type 1 IFN, as well as induces pyroptosis [
129,
130,
131]. The AIM2-induced GSDMD functions as a negative regulator of type 1 interferon production mediated by cGAS-STING [
125]. In addition, the AIM2-ASC inflammasome inhibits the STING-TBK1 (TRAF family member-associated NF-κB activator-binding kinase 1) interaction required for the IRF3-dependent release of type 1 interferons [
131,
132]. In the absence of certain cytosolic DNAs, AIM2 remains inactive [
132]. A schematic representation of the cfDNA recognition and consequent pathway activation is shown in
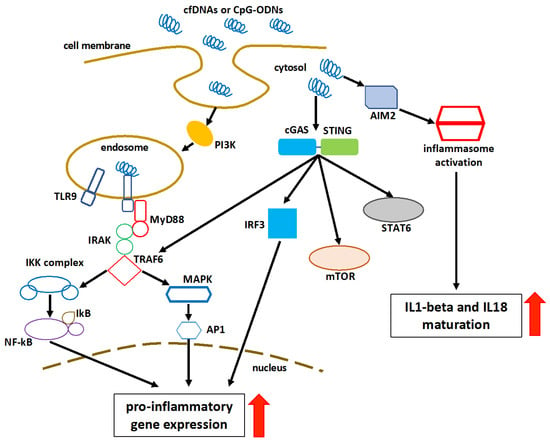
Figure 2.
Figure 2. Schematic representation of cell-free DNA sensing and consequent pathway activation. Class III PI3K promotes the internalization of cfDNA and CpG-ODNs into TLR9-containing endosomal vesicles. The intracytoplasmic activation signal is transmitted via the interaction of cfDNA and TLR9. MyD88 is recruited to the Toll–interleukin-1 receptor domain of TLR9, followed by the activation of the IRAK–TRAF6 complex. This activates both the MAPK and the inhibitor of IKK complexes, resulting in the overexpression of transcription factors, such as NF-kB and AP1. cGAS-mediated detection of cytosolic DNA initiates a STING-dependent reaction. The cGAS-STING pathway can also activate IRFs, mTOR, STAT6, and MAPK in a direct or indirect way. In the cytosol, AIM2 binds to the double-stranded DNA, resulting in the creation of the AIM2 inflammasome. This results in the activation of caspase 1, the maturation of proinflammatory cytokines IL-1β and IL-18, and finally pyroptosis. AP1: activator protein 1; red arrows: upregulation.