Autophagy is a stress-induced process that eliminates damaged organelles and dysfunctional cargos in cytoplasm, including unfolded proteins. Autophagy is involved in constructing the immunosuppressive microenvironment during tumor initiation and progression. It appears to be one of the most common processes involved in cancer immunotherapy, playing bidirectional roles in immunotherapy. Accumulating evidence suggests that inducing or inhibiting autophagy contributes to immunotherapy efficacy. Exploring autophagy targets and their modifiers to control autophagy in the tumor microenvironment is an emerging strategy to facilitate cancer immunotherapy.
- autophagy
- cancer
- immunotherapy
1. Introduction
2. Autophagy in Immune Cells
2.1. Autophagy in T cells
2.2. Autophagy in B cells
2.3. Autophagy in Natural Killer (NK) Cells
2.4. Autophagy in Dendritic Cells (DCs)
2.5. Autophagy in Macrophages
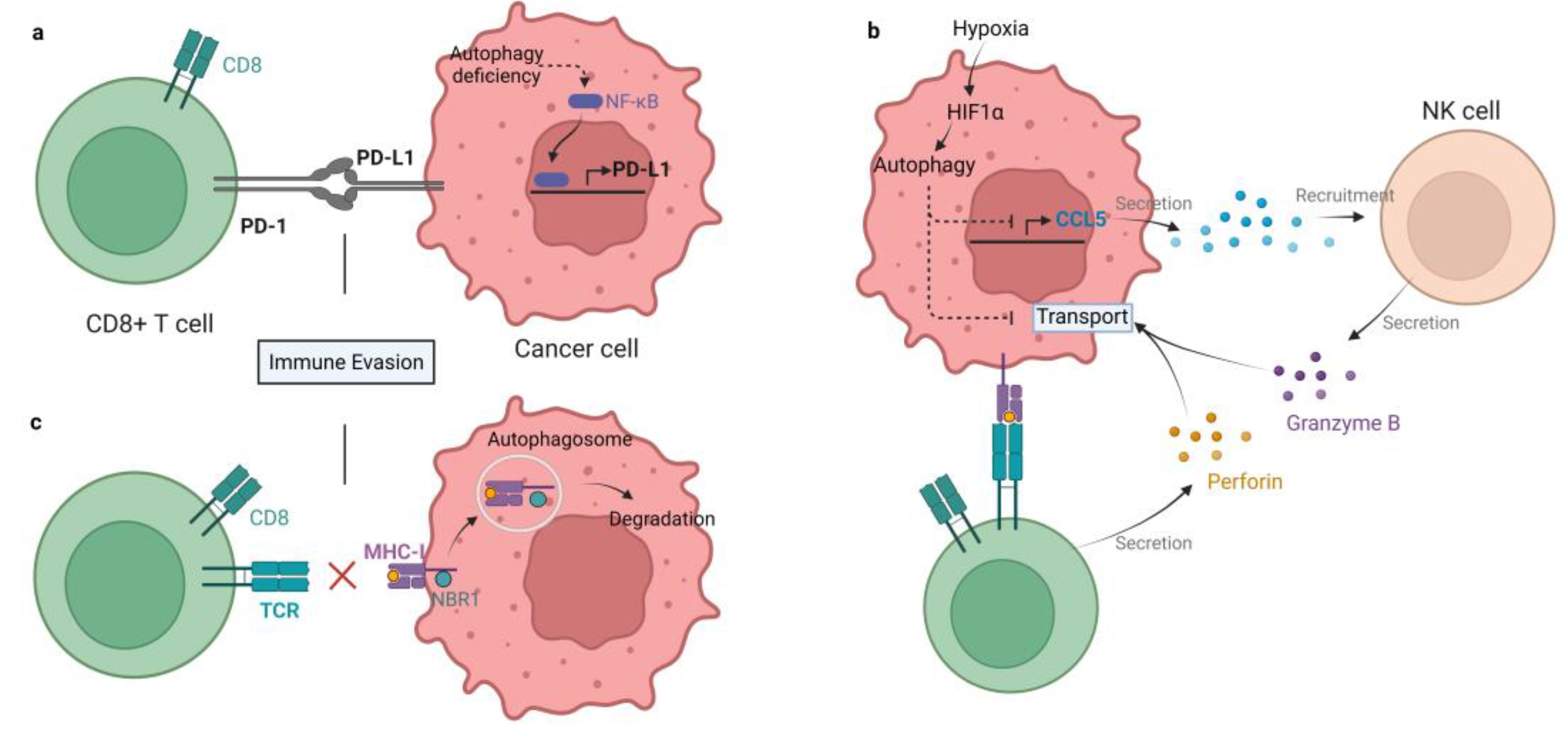
3. Autophagy in Regulating Immune Checkpoint Molecules
4. Autophagy in Immune Cytokines
4.1. Interleukins
4.2. Interferons
4.3. Transforming Growth Factor Beta (TGF-β)
4.4. Tumor Necrosis Factor Alpha (TNF-α)
This entry is adapted from the peer-reviewed paper 10.3390/cells11192996
References
- Ma, Y.; Galluzzi, L.; Zitvogel, L.; Kroemer, G. Autophagy and cellular immune responses. Immunity 2013, 39, 211–227.
- Zhan, Z.; Xie, X.; Cao, H.; Zhou, X.; Zhang, X.D.; Fan, H.; Liu, Z. Autophagy facilitates TLR4- and TLR3-triggered migration and invasion of lung cancer cells through the promotion of TRAF6 ubiquitination. Autophagy 2014, 10, 257–268.
- Tang, D.; Kang, R.; Cheh, C.W.; Livesey, K.M.; Liang, X.; Schapiro, N.E.; Benschop, R.; Sparvero, L.J.; Amoscato, A.A.; Tracey, K.J.; et al. HMGB1 release and redox regulates autophagy and apoptosis in cancer cells. Oncogene 2010, 29, 5299–5310.
- Schmeisser, H.; Fey, S.B.; Horowitz, J.; Fischer, E.R.; Balinsky, C.A.; Miyake, K.; Bekisz, J.; Snow, A.L.; Zoon, K.C. Type I interferons induce autophagy in certain human cancer cell lines. Autophagy 2013, 9, 683–696.
- Kiyono, K.; Suzuki, H.I.; Matsuyama, H.; Morishita, Y.; Komuro, A.; Kano, M.R.; Sugimoto, K.; Miyazono, K. Autophagy is activated by TGF-beta and potentiates TGF-beta-mediated growth inhibition in human hepatocellular carcinoma cells. Cancer Res. 2009, 69, 8844–8852.
- Katheder, N.S.; Khezri, R.; O’Farrell, F.; Schultz, S.W.; Jain, A.; Rahman, M.M.; Schink, K.O.; Theodossiou, T.A.; Johansen, T.; Juhasz, G.; et al. Microenvironmental autophagy promotes tumour growth. Nature 2017, 541, 417–420.
- Zou, W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat. Rev. Cancer 2005, 5, 263–274.
- Xia, H.; Green, D.R.; Zou, W. Autophagy in tumour immunity and therapy. Nat. Rev. Cancer 2021, 21, 281–297.
- Semmling, V.; Lukacs-Kornek, V.; Thaiss, C.A.; Quast, T.; Hochheiser, K.; Panzer, U.; Rossjohn, J.; Perlmutter, P.; Cao, J.; Godfrey, D.I.; et al. Alternative cross-priming through CCL17-CCR4-mediated attraction of CTLs toward NKT cell-licensed DCs. Nat. Immunol. 2010, 11, 313–320.
- Li, H.; Li, Y.; Jiao, J.; Hu, H.M. Alpha-alumina nanoparticles induce efficient autophagy-dependent cross-presentation and potent antitumour response. Nat. Nanotechnol. 2011, 6, 645–650.
- Yang, S.; Imamura, Y.; Jenkins, R.W.; Canadas, I.; Kitajima, S.; Aref, A.; Brannon, A.; Oki, E.; Castoreno, A.; Zhu, Z.; et al. Autophagy Inhibition Dysregulates TBK1 Signaling and Promotes Pancreatic Inflammation. Cancer Immunol. Res. 2016, 4, 520–530.
- Wang, X.; Wu, W.K.K.; Gao, J.; Li, Z.; Dong, B.; Lin, X.; Li, Y.; Li, Y.; Gong, J.; Qi, C.; et al. Autophagy inhibition enhances PD-L1 expression in gastric cancer. J. Exp. Clin. Cancer Res. 2019, 38, 140.
- Deretic, V.; Levine, B. Autophagy balances inflammation in innate immunity. Autophagy 2018, 14, 243–251.
- Mathew, R.; Khor, S.; Hackett, S.R.; Rabinowitz, J.D.; Perlman, D.H.; White, E. Functional role of autophagy-mediated proteome remodeling in cell survival signaling and innate immunity. Mol. Cell 2014, 55, 916–930.
- Poillet-Perez, L.; Sharp, D.W.; Yang, Y.; Laddha, S.V.; Ibrahim, M.; Bommareddy, P.K.; Hu, Z.S.; Vieth, J.; Haas, M.; Bosenberg, M.W.; et al. Autophagy promotes growth of tumors with high mutational burden by inhibiting a T-cell immune response. Nat. Cancer 2020, 1, 923–934.
- Yatim, N.; Cullen, S.; Albert, M.L. Dying cells actively regulate adaptive immune responses. Nat. Rev. Immunol. 2017, 17, 262–275.
- DeVorkin, L.; Pavey, N.; Carleton, G.; Comber, A.; Ho, C.; Lim, J.; McNamara, E.; Huang, H.; Kim, P.; Zacharias, L.G.; et al. Autophagy Regulation of Metabolism Is Required for CD8(+) T Cell Anti-tumor Immunity. Cell Rep. 2019, 27, 502–513.e5.
- Yamamoto, K.; Venida, A.; Yano, J.; Biancur, D.E.; Kakiuchi, M.; Gupta, S.; Sohn, A.S.W.; Mukhopadhyay, S.; Lin, E.Y.; Parker, S.J.; et al. Autophagy promotes immune evasion of pancreatic cancer by degrading MHC-I. Nature 2020, 581, 100–105.
- Garg, A.D.; Dudek, A.M.; Agostinis, P. Autophagy-dependent suppression of cancer immunogenicity and effector mechanisms of innate and adaptive immunity. Oncoimmunology 2013, 2, e26260.
- Baginska, J.; Viry, E.; Berchem, G.; Poli, A.; Noman, M.Z.; van Moer, K.; Medves, S.; Zimmer, J.; Oudin, A.; Niclou, S.P.; et al. Granzyme B degradation by autophagy decreases tumor cell susceptibility to natural killer-mediated lysis under hypoxia. Proc. Natl. Acad. Sci. USA 2013, 110, 17450–17455.
- Noman, M.Z.; Janji, B.; Kaminska, B.; Van Moer, K.; Pierson, S.; Przanowski, P.; Buart, S.; Berchem, G.; Romero, P.; Mami-Chouaib, F.; et al. Blocking hypoxia-induced autophagy in tumors restores cytotoxic T-cell activity and promotes regression. Cancer Res. 2011, 71, 5976–5986.
- Zhong, Z.; Sanchez-Lopez, E.; Karin, M. Autophagy, Inflammation, and Immunity: A Troika Governing Cancer and Its Treatment. Cell 2016, 166, 288–298.
- Blum, J.S.; Wearsch, P.A.; Cresswell, P. Pathways of antigen processing. Annu. Rev. Immunol. 2013, 31, 443–473.
- Loi, M.; Muller, A.; Steinbach, K.; Niven, J.; Barreira da Silva, R.; Paul, P.; Ligeon, L.A.; Caruso, A.; Albrecht, R.A.; Becker, A.C.; et al. Macroautophagy Proteins Control MHC Class I Levels on Dendritic Cells and Shape Anti-viral CD8(+) T Cell Responses. Cell Rep. 2016, 15, 1076–1087.
- Parekh, V.V.; Pabbisetty, S.K.; Wu, L.; Sebzda, E.; Martinez, J.; Zhang, J.; Van Kaer, L. Autophagy-related protein Vps34 controls the homeostasis and function of antigen cross-presenting CD8alpha(+) dendritic cells. Proc. Natl. Acad. Sci. USA 2017, 114, E6371–E6380.
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (B7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv324.
- Jang, Y.J.; Kim, J.H.; Byun, S. Modulation of Autophagy for Controlling Immunity. Cells 2019, 8, 138.
- Clarke, A.J.; Simon, A.K. Autophagy in the renewal, differentiation and homeostasis of immune cells. Nat. Rev. Immunol. 2019, 19, 170–183.
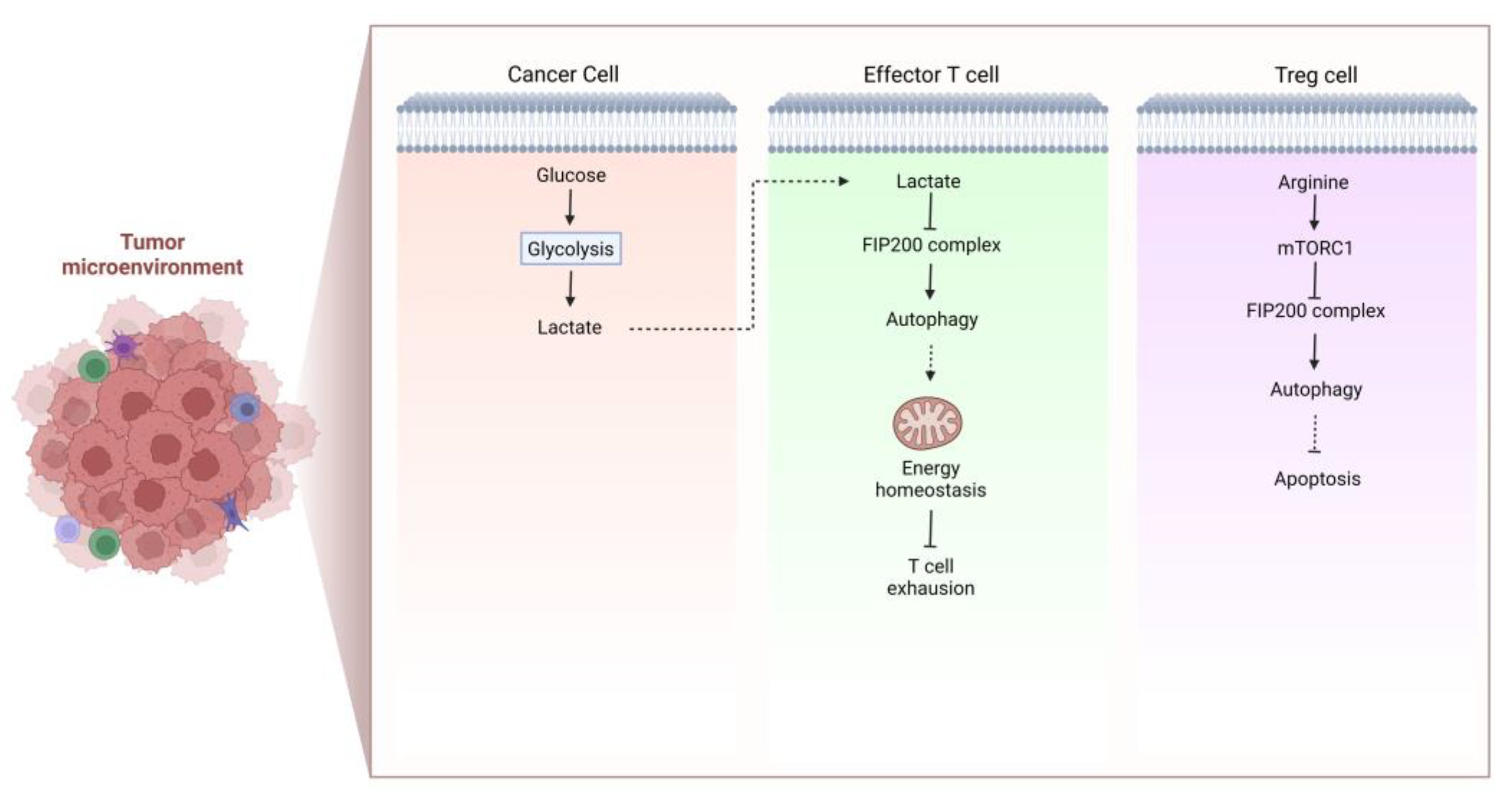
- Xia, H.; Wang, W.; Crespo, J.; Kryczek, I.; Li, W.; Wei, S.; Bian, Z.; Maj, T.; He, M.; Liu, R.J.; et al. Suppression of FIP200 and autophagy by tumor-derived lactate promotes naive T cell apoptosis and affects tumor immunity. Sci. Immunol. 2017, 2, eaan4631.
- Sena, L.A.; Li, S.; Jairaman, A.; Prakriya, M.; Ezponda, T.; Hildeman, D.A.; Wang, C.R.; Schumacker, P.T.; Licht, J.D.; Perlman, H.; et al. Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 2013, 38, 225–236.
- Jia, W.; He, M.X.; McLeod, I.X.; Guo, J.; Ji, D.; He, Y.W. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy 2015, 11, 2335–2345.
- Bronietzki, A.W.; Schuster, M.; Schmitz, I. Autophagy in T-cell development, activation and differentiation. Immunol. Cell Biol. 2015, 93, 25–34.
- Yang, G.; Song, W.; Postoak, J.L.; Chen, J.; Martinez, J.; Zhang, J.; Wu, L.; Van Kaer, L. Autophagy-related protein PIK3C3/VPS34 controls T cell metabolism and function. Autophagy 2021, 17, 1193–1204.
- Pua, H.H.; Guo, J.; Komatsu, M.; He, Y.W. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J. Immunol. 2009, 182, 4046–4055.
- Swadling, L.; Pallett, L.J.; Diniz, M.O.; Baker, J.M.; Amin, O.E.; Stegmann, K.A.; Burton, A.R.; Schmidt, N.M.; Jeffery-Smith, A.; Zakeri, N.; et al. Human Liver Memory CD8(+) T Cells Use Autophagy for Tissue Residence. Cell Rep. 2020, 30, 687–698.e686.
- Delgoffe, G.M.; Kole, T.P.; Zheng, Y.; Zarek, P.E.; Matthews, K.L.; Xiao, B.; Worley, P.F.; Kozma, S.C.; Powell, J.D. The mTOR kinase differentially regulates effector and regulatory T cell lineage commitment. Immunity 2009, 30, 832–844.
- Le Texier, L.; Lineburg, K.E.; Cao, B.; McDonald-Hyman, C.; Leveque-El Mouttie, L.; Nicholls, J.; Melino, M.; Nalkurthi, B.C.; Alexander, K.A.; Teal, B.; et al. Autophagy-dependent regulatory T cells are critical for the control of graft-versus-host disease. JCI Insight 2016, 1, e86850.
- Wei, J.; Long, L.; Yang, K.; Guy, C.; Shrestha, S.; Chen, Z.; Wu, C.; Vogel, P.; Neale, G.; Green, D.R.; et al. Autophagy enforces functional integrity of regulatory T cells by coupling environmental cues and metabolic homeostasis. Nat. Immunol. 2016, 17, 277–285.
- Becher, J.; Simula, L.; Volpe, E.; Procaccini, C.; La Rocca, C.; D’Acunzo, P.; Cianfanelli, V.; Strappazzon, F.; Caruana, I.; Nazio, F.; et al. AMBRA1 Controls Regulatory T-Cell Differentiation and Homeostasis Upstream of the FOXO3-FOXP3 Axis. Dev. Cell 2018, 47, 592–607.e596.
- Munz, C. Autophagy proteins in antigen processing for presentation on MHC molecules. Immunol. Rev. 2016, 272, 17–27.
- Li, W.; Tanikawa, T.; Kryczek, I.; Xia, H.; Li, G.; Wu, K.; Wei, S.; Zhao, L.; Vatan, L.; Wen, B.; et al. Aerobic Glycolysis Controls Myeloid-Derived Suppressor Cells and Tumor Immunity via a Specific CEBPB Isoform in Triple-Negative Breast Cancer. Cell Metab. 2018, 28, 87–103.e106.
- Parker, K.H.; Horn, L.A.; Ostrand-Rosenberg, S. High-mobility group box protein 1 promotes the survival of myeloid-derived suppressor cells by inducing autophagy. J. Leukoc. Biol. 2016, 100, 463–470.
- Alissafi, T.; Hatzioannou, A.; Mintzas, K.; Barouni, R.M.; Banos, A.; Sormendi, S.; Polyzos, A.; Xilouri, M.; Wielockx, B.; Gogas, H.; et al. Autophagy orchestrates the regulatory program of tumor-associated myeloid-derived suppressor cells. J. Clin. Investig. 2018, 128, 3840–3852.
- Pan, H.; Chen, L.; Xu, Y.; Han, W.; Lou, F.; Fei, W.; Liu, S.; Jing, Z.; Sui, X. Autophagy-associated immune responses and cancer immunotherapy. Oncotarget 2016, 7, 21235–21246.
- Arnold, J.; Murera, D.; Arbogast, F.; Fauny, J.D.; Muller, S.; Gros, F. Autophagy is dispensable for B-cell development but essential for humoral autoimmune responses. Cell Death Differ. 2016, 23, 853–864.
- Fribourg, M.; Ni, J.; Nina Papavasiliou, F.; Yue, Z.; Heeger, P.S.; Leventhal, J.S. Allospecific Memory B Cell Responses Are Dependent on Autophagy. Am. J. Transpl. 2018, 18, 102–112.
- Wang, S.; Xia, P.; Huang, G.; Zhu, P.; Liu, J.; Ye, B.; Du, Y.; Fan, Z. FoxO1-mediated autophagy is required for NK cell development and innate immunity. Nat. Commun. 2016, 7, 11023.
- Salio, M.; Puleston, D.J.; Mathan, T.S.; Shepherd, D.; Stranks, A.J.; Adamopoulou, E.; Veerapen, N.; Besra, G.S.; Hollander, G.A.; Simon, A.K.; et al. Essential role for autophagy during invariant NKT cell development. Proc. Natl. Acad. Sci. USA 2014, 111, E5678–E5687.
- Pei, B.; Zhao, M.; Miller, B.C.; Vela, J.L.; Bruinsma, M.W.; Virgin, H.W.; Kronenberg, M. Invariant NKT cells require autophagy to coordinate proliferation and survival signals during differentiation. J. Immunol. 2015, 194, 5872–5884.
- Liu, E.; Van Grol, J.; Subauste, C.S. Atg5 but not Atg7 in dendritic cells enhances IL-2 and IFN-gamma production by Toxoplasma gondii-reactive CD4+ T cells. Microbes Infect. 2015, 17, 275–284.
- Seto, S.; Tsujimura, K.; Horii, T.; Koide, Y. Autophagy adaptor protein p62/SQSTM1 and autophagy-related gene Atg5 mediate autophagosome formation in response to Mycobacterium tuberculosis infection in dendritic cells. PLoS ONE 2013, 8, e86017.
- Lee, H.K.; Mattei, L.M.; Steinberg, B.E.; Alberts, P.; Lee, Y.H.; Chervonsky, A.; Mizushima, N.; Grinstein, S.; Iwasaki, A. In vivo requirement for Atg5 in antigen presentation by dendritic cells. Immunity 2010, 32, 227–239.
- Chen, P.; Cescon, M.; Bonaldo, P. Autophagy-mediated regulation of macrophages and its applications for cancer. Autophagy 2014, 10, 192–200.
- Jacquel, A.; Obba, S.; Solary, E.; Auberger, P. Proper macrophagic differentiation requires both autophagy and caspase activation. Autophagy 2012, 8, 1141–1143.
- Zhang, Y.; Morgan, M.J.; Chen, K.; Choksi, S.; Liu, Z.G. Induction of autophagy is essential for monocyte-macrophage differentiation. Blood 2012, 119, 2895–2905.
- Liu, K.; Zhao, E.; Ilyas, G.; Lalazar, G.; Lin, Y.; Haseeb, M.; Tanaka, K.E.; Czaja, M.J. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy 2015, 11, 271–284.
- Li, N.; Qin, J.; Lan, L.; Zhang, H.; Liu, F.; Wu, Z.; Ni, H.; Wang, Y. PTEN inhibits macrophage polarization from M1 to M2 through CCL2 and VEGF-A reduction and NHERF-1 synergism. Cancer Biol. Ther. 2015, 16, 297–306.
- Khatoon, E.; Parama, D.; Kumar, A.; Alqahtani, M.S.; Abbas, M.; Girisa, S.; Sethi, G.; Kunnumakkara, A.B. Targeting PD-1/PD-L1 axis as new horizon for ovarian cancer therapy. Life Sci. 2022, 306, 120827.
- Robainas, M.; Otano, R.; Bueno, S.; Ait-Oudhia, S. Understanding the role of PD-L1/PD1 pathway blockade and autophagy in cancer therapy. OncoTargets Ther. 2017, 10, 1803–1807.
- Maher, C.M.; Thomas, J.D.; Haas, D.A.; Longen, C.G.; Oyer, H.M.; Tong, J.Y.; Kim, F.J. Small-Molecule Sigma1 Modulator Induces Autophagic Degradation of PD-L1. Mol. Cancer Res. 2018, 16, 243–255.
- Clark, C.A.; Gupta, H.B.; Curiel, T.J. Tumor cell-intrinsic CD274/PD-L1: A novel metabolic balancing act with clinical potential. Autophagy 2017, 13, 987–988.
- Shukla, S.A.; Bachireddy, P.; Schilling, B.; Galonska, C.; Zhan, Q.; Bango, C.; Langer, R.; Lee, P.C.; Gusenleitner, D.; Keskin, D.B.; et al. Cancer-Germline Antigen Expression Discriminates Clinical Outcome to CTLA-4 Blockade. Cell 2018, 173, 624–633.e628.
- Kato, H.; Perl, A. Blockade of Treg Cell Differentiation and Function by the Interleukin-21-Mechanistic Target of Rapamycin Axis Via Suppression of Autophagy in Patients With Systemic Lupus Erythematosus. Arthritis Rheumatol. 2018, 70, 427–438.
- Alissafi, T.; Banos, A.; Boon, L.; Sparwasser, T.; Ghigo, A.; Wing, K.; Vassilopoulos, D.; Boumpas, D.; Chavakis, T.; Cadwell, K.; et al. Tregs restrain dendritic cell autophagy to ameliorate autoimmunity. J. Clin. Investig. 2017, 127, 2789–2804.
- Folgiero, V.; Miele, E.; Carai, A.; Ferretti, E.; Alfano, V.; Po, A.; Bertaina, V.; Goffredo, B.M.; Benedetti, M.C.; Camassei, F.D.; et al. IDO1 involvement in mTOR pathway: A molecular mechanism of resistance to mTOR targeting in medulloblastoma. Oncotarget 2016, 7, 52900–52911.
- Metz, R.; Rust, S.; Duhadaway, J.B.; Mautino, M.R.; Munn, D.H.; Vahanian, N.N.; Link, C.J.; Prendergast, G.C. IDO inhibits a tryptophan sufficiency signal that stimulates mTOR: A novel IDO effector pathway targeted by D-1-methyl-tryptophan. Oncoimmunology 2012, 1, 1460–1468.
- McGaha, T.L. IDO-GCN2 and autophagy in inflammation. Oncotarget 2015, 6, 21771–21772.
- Schauer, I.G.; Zhang, J.; Xing, Z.; Guo, X.; Mercado-Uribe, I.; Sood, A.K.; Huang, P.; Liu, J. Interleukin-1beta promotes ovarian tumorigenesis through a p53/NF-kappaB-mediated inflammatory response in stromal fibroblasts. Neoplasia 2013, 15, 409–420.
- Jiang, S.; Dupont, N.; Castillo, E.F.; Deretic, V. Secretory versus degradative autophagy: Unconventional secretion of inflammatory mediators. J. Innate Immun. 2013, 5, 471–479.
- Peral de Castro, C.; Jones, S.A.; Ni Cheallaigh, C.; Hearnden, C.A.; Williams, L.; Winter, J.; Lavelle, E.C.; Mills, K.H.; Harris, J. Autophagy regulates IL-23 secretion and innate T cell responses through effects on IL-1 secretion. J. Immunol. 2012, 189, 4144–4153.
- Sun, K.; Xu, L.; Jing, Y.; Han, Z.; Chen, X.; Cai, C.; Zhao, P.; Zhao, X.; Yang, L.; Wei, L. Autophagy-deficient Kupffer cells promote tumorigenesis by enhancing mtROS-NF-kappaB-IL1alpha/beta-dependent inflammation and fibrosis during the preneoplastic stage of hepatocarcinogenesis. Cancer Lett. 2017, 388, 198–207.
- Kang, R.; Tang, D.; Lotze, M.T.; Zeh Iii, H.J. Autophagy is required for IL-2-mediated fibroblast growth. Exp. Cell Res. 2013, 319, 556–565.
- Liang, X.; De Vera, M.E.; Buchser, W.J.; Romo de Vivar Chavez, A.; Loughran, P.; Beer Stolz, D.; Basse, P.; Wang, T.; Van Houten, B.; Zeh, H.J., 3rd; et al. Inhibiting systemic autophagy during interleukin 2 immunotherapy promotes long-term tumor regression. Cancer Res. 2012, 72, 2791–2801.
- Qin, B.; Zhou, Z.; He, J.; Yan, C.; Ding, S. IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci. Rep. 2015, 5, 15701.
- Linnemann, A.K.; Blumer, J.; Marasco, M.R.; Battiola, T.J.; Umhoefer, H.M.; Han, J.Y.; Lamming, D.W.; Davis, D.B. Interleukin 6 protects pancreatic beta cells from apoptosis by stimulation of autophagy. FASEB J. 2017, 31, 4140–4152.
- Santarelli, R.; Gonnella, R.; Di Giovenale, G.; Cuomo, L.; Capobianchi, A.; Granato, M.; Gentile, G.; Faggioni, A.; Cirone, M. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci. Rep. 2014, 4, 4241.
- Cho, S.H.; Oh, S.Y.; Lane, A.P.; Lee, J.; Oh, M.H.; Lee, S.; Zheng, T.; Zhu, Z. Regulation of nasal airway homeostasis and inflammation in mice by SHP-1 and Th2/Th1 signaling pathways. PLoS ONE 2014, 9, e103685.
- Qi, G.M.; Jia, L.X.; Li, Y.L.; Li, H.H.; Du, J. Adiponectin suppresses angiotensin II-induced inflammation and cardiac fibrosis through activation of macrophage autophagy. Endocrinology 2014, 155, 2254–2265.
- Schmeisser, H.; Bekisz, J.; Zoon, K.C. New function of type I IFN: Induction of autophagy. J. Interf. Cytokine Res. 2014, 34, 71–78.
- Buchser, W.J.; Laskow, T.C.; Pavlik, P.J.; Lin, H.M.; Lotze, M.T. Cell-mediated autophagy promotes cancer cell survival. Cancer Res. 2012, 72, 2970–2979.
- Matsuzawa, T.; Kim, B.H.; Shenoy, A.R.; Kamitani, S.; Miyake, M.; Macmicking, J.D. IFN-gamma elicits macrophage autophagy via the p38 MAPK signaling pathway. J. Immunol. 2012, 189, 813–818.
- Tu, S.P.; Quante, M.; Bhagat, G.; Takaishi, S.; Cui, G.; Yang, X.D.; Muthuplani, S.; Shibata, W.; Fox, J.G.; Pritchard, D.M.; et al. IFN-gamma inhibits gastric carcinogenesis by inducing epithelial cell autophagy and T-cell apoptosis. Cancer Res. 2011, 71, 4247–4259.
- Hubbard, V.M.; Valdor, R.; Patel, B.; Singh, R.; Cuervo, A.M.; Macian, F. Macroautophagy regulates energy metabolism during effector T cell activation. J. Immunol. 2010, 185, 7349–7357.
- Wilson, E.B.; El-Jawhari, J.J.; Neilson, A.L.; Hall, G.D.; Melcher, A.A.; Meade, J.L.; Cook, G.P. Human tumour immune evasion via TGF-beta blocks NK cell activation but not survival allowing therapeutic restoration of anti-tumour activity. PLoS ONE 2011, 6, e22842.
- Ding, Y.; Kim, S.; Lee, S.Y.; Koo, J.K.; Wang, Z.; Choi, M.E. Autophagy regulates TGF-beta expression and suppresses kidney fibrosis induced by unilateral ureteral obstruction. J. Am. Soc. Nephrol. 2014, 25, 2835–2846.
- Zhang, C.; Zhang, X.; Xu, R.; Huang, B.; Chen, A.J.; Li, C.; Wang, J.; Li, X.G. TGF-beta2 initiates autophagy via Smad and non-Smad pathway to promote glioma cells’ invasion. J. Exp. Clin. Cancer Res. 2017, 36, 162.
- Suzuki, H.I.; Kiyono, K.; Miyazono, K. Regulation of autophagy by transforming growth factor-beta (TGF-beta) signaling. Autophagy 2010, 6, 645–647.
- Wang, M.X.; Cheng, X.Y.; Jin, M.; Cao, Y.L.; Yang, Y.P.; Wang, J.D.; Li, Q.; Wang, F.; Hu, L.F.; Liu, C.F. TNF compromises lysosome acidification and reduces alpha-synuclein degradation via autophagy in dopaminergic cells. Exp. Neurol. 2015, 271, 112–121.
- Ullio, C.; Brunk, U.T.; Urani, C.; Melchioretto, P.; Bonelli, G.; Baccino, F.M.; Autelli, R. Autophagy of metallothioneins prevents TNF-induced oxidative stress and toxicity in hepatoma cells. Autophagy 2015, 11, 2184–2198.
- Pun, N.T.; Subedi, A.; Kim, M.J.; Park, P.H. Globular Adiponectin Causes Tolerance to LPS-Induced TNF-alpha Expression via Autophagy Induction in RAW 264.7 Macrophages: Involvement of SIRT1/FoxO3A Axis. PLoS ONE 2015, 10, e0124636.