Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Agronomy
The present paper focus on the recent genetic and genome-based research findings in the allelopathic field, with a particular emphasis on weed control, which is one of the major crop yield limiting factors. The key plant-microorganism interactions including the cross-Kingdome RNAi phenomenon and the involvement of microRNAs in allelopathy are discussed. Through this review we wanted to put the foundation for advancing knowledge in allelopathy and uncover the areas where research is needed.
- allelopathic genes
- quantitative trait loci (QTL)
- weed control
- microorganisms
- Next Generation Sequencing
- Cross-kingdome RNAi
1. Introduction
Allelopathy is an ecological phenomenon, in which the chemicals produced by plants and microorganisms affect the growth, development, and fitness of other organisms [1]. This discipline represents a topic of growing interest due to the sustainability discussion currently in progress [2]. Over the years, several definitions have been adopted, in which “interaction” has been the key common word. Many definitions of allelopathy have been given throughout history [3,4,5]. More recently, the International Allelopathy Society (IAS) has further expanded the definition as follows: “any process involving secondary metabolites produced by plants, microorganisms, viruses, and fungi that influence the growth and development of agricultural and biological systems” (IAS, 1996) (Figure 1). However, although the different definitions mentioned above have tried to include all the possible physiological responses due to allelopathic interactions induced by secondary metabolites among organisms, to date the positive or negative effects of allelopathy are not well defined [5].
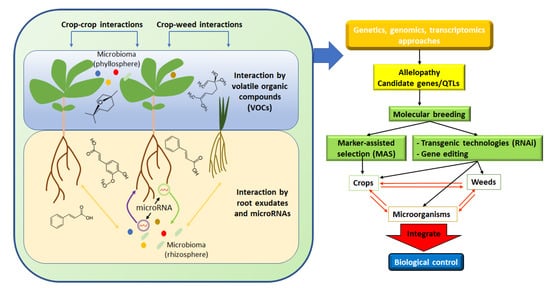
Figure 1. Schematic diagram highlighting the different allelopathic interactions. Aboveground interactions are mediated by volatile organic compounds (VOCs), whereas root exudates represent the main factor mediating allelopathic interaction in the soil. Two strategies (transgenic organisms or MAS) can be pursued to create new varieties with higher allelopathic potential. Microbiota modulates, reduces, or enhances, allelopathic interactions even through cross-kingdom microRNA exchanges.
Indeed, the study of plant responses to allelochemicals is markedly influenced by the used methods, the considered biological traits, and the evolutionary history of the organisms [5]. In addition, the allelopathic phenomenon increases with the genetic distance between the species, and this concept opens a new scenario in which kin recognition takes place among species [6] and where root exudates play a pivotal role [7]. In this context, Crepy and Casal [8] described for the first time the molecular mechanisms of recognition responses in the shoot in which phytochrome B and cryptochrome 1 genes were involved. Moreover, plants can be passive organisms, responding only to environmental fluctuations, or active, transmitting, receiving, and reacting directly with other plants and microorganisms to chemical signals, regardless of environmental variations (Figure 1) [9]. These responses, which determine a fundamental role in the acquisition of resources, are the key to how a plant community is organized and how species-specific mechanisms, such as coevolution, are modulated [10,11,12].
In field conditions, the allelopathic phenomenon can also be easily understood from the spatial distribution of species [13,14,15,16,17]. taking into account the architecture of the root system as well [18]. Moreover, for many years, allelopathy was considered an aspect of the plant competition phenomenon, but today, the distinction between the two phenomena is very clear [19,20].
Allelochemicals are secondary metabolites from different classes, such as phenolic, terpenoid, and alkaloid compounds [21]. As the main allelochemicals within plants, in terms of proportion, the phenolic compounds were extensively studied to identify their allelopathic mechanism of action in model species, such as Arabidopsis thaliana and Lactuca sativa, although no field applications have been performed [21]. Several studies reported the allelopathic effect of simple phenolic compounds on the morphophysiological processes in many crops, such as root morphology in maize plants [22], membrane permeability [23], nutrient uptake [22], cell division and elongation [24], photosynthesis and respiration [25], and hormones synthesis and balance [26].
On the other hand, more complex metabolites were also studied in allelopathic interaction such as terpenoids, including mono-, di-, triterpenoids, and sterols [21], which are involved in seed germination and oxidative damage [26], plant communication with other organisms [27], and plant defense as well [28].
Moreover, in agroecosystems, the allelopathic effects take on considerable importance in weed management [29]. Indeed, among crop pests, agricultural weeds represent the major limitation to agricultural production [30], and chemical weed control currently represents the most adopted strategy, leading to environment and human health cues [31]. A strategy to overcome these problems could be the use of allelochemicals, which possess a high potential as bioherbicide or/and herbicide bioinspired, exploiting new mechanisms of action, thereby overcoming specific resistances [12], thus representing an alternative to weed control in terms of sustainability.
2. Genomic Approaches in Allelopathy
In recent years, genetic technologies have developed rapidly, and whole-genome sequencing has become an increasingly routine technique in many areas, such as medicine, biotechnology, and agriculture. Although global expression responses of plant genomes in allelopathy using DNA microarrays were reported [37], the new technologies could detect novel transcripts, and predict the gene regulatory networks of a biological response. Indeed, to analyze gene and/or regulatory chromosome regions, a complete sequence assembly is necessary, and, at the same time, the biological processes associated with likely phenotypic traits can be determined. To date, these sequencing technologies, such as whole-genome sequencing and RNA sequencing (RNA-seq), can be performed at different biological levels: from plant tissue to single cell [38]. However, in allelopathy, the application of these approaches is markedly limited to a few experiments (Table 1), including either gene expression or RNA sequencing technologies, as well as the identification of quantitative trait loci (QTL) useful for plant breeding.
Table 1. Representative articles of the genetic and genomic approaches in allelopathy.
| Plant Material | Methods | Targets | References |
|---|---|---|---|
| Common reed | RNA-seq | Phytohormones | He et al. [39] |
| Rice/barnyard grass | Microarray | Phytohormones | Chi et al. [40] |
| Tomato | RNA-seq | Antioxidants and Hormones | Cheng et al. [34] |
| Rice/barnyard grass | RNA-seq | Shikimic acid and acetic acid pathways | Zhang et al. [41] |
| Rice/barnyard grass | RNA-seq | Diterpenoid and flavonoid biosynthesis pathway | Li et al. [42] |
| Rehmannia glutinosa | Cloning, qRT-PCR | Phenolic biosynthesis: C3H gene | Yang et al. [43] |
| Soybeans | RNA-seq | Oxidative stress and jasmonic acid signaling (PIF3) |
Horvath et al. [44] |
| Rice | SNPs genotyping | QTL regions | Chung et al. [45] |
| Rice | qRT-PCR | Biosynthesis of phenolic acids | Zhang et al. [46] |
| Wheat/ryegrass | AFLP, RFLP and SSR genotyping | QTL regions | Wu et al. [47] |
| Lettuce/rice | RFLP genotyping | QTL regions | Zeng et al. [48] |
| Lettuce/Triticum Speltoides | RAPD genotyping | Genetic diversity in allelopathic potential | Quader et al. [49] |
| Rice | RNA interference | PAL gene expression | Fang et al. [50] |
| Rice | T-DNA insertion | OsCPS4, OsKSL4 | Xu et al. [51] |
| Rice/barnyard grass | qRT-PCR | PAL, C4H, F5H, and COMT genes | Zhang et al. [52] |
| Sorghum | SSR genotyping | QTL regions | Shehzad et al. [53] |
| Rice/barnyard grass | qRT-PCR, ChIP-seq, ChIP-qPCR | MYB transcription factor | Fang et al. [35] |
| Rice | qRT-PCR, RNA-seq | Biosynthetic gene clusters | Sultana et al. [54] |
| Arabidopsis | RNA-seq | Signal transduction, nutrient transporter, detoxification genes | Zhang et al. [36] |
| Rice | RNA-seq | Chlorophyll and nitrogen metabolisms | Li et al. [55] |
| Rice | Genome sequencing | detoxification-related genes (CYP450, GST) DIMBOA gene cluster. | Guo et al. [56] |
2.1. From Metabolite to Gene in Plants
Despite the considerable number of manuscripts dealing with crop allelopathy, the main works aiming to understand the molecular networks involved in allelopathic traits and their potential involvement in breeding programs for field application were performed on cereals (Table 1). In particular, the main metabolites considered to be involved in this phenomenon belong to the classes of indoles (benzoxazinoids and their derivatives, produced by wheat, maize, rice, etc.), phenylpropanoids (i.e., cinnamic acids derivatives), and terpenoids (momilactones a and b, produced by rice).
Among the molecular approaches used to clarify some aspects related to the allelopathic phenomenon, the expression analyses of plant secondary metabolism pathway-related genes turned out to be an informative technique for the regulation of metabolite biosynthesis. The first correlation between metabolite produced and gene expression was demonstrated in cereals [50,57]. In particular, some studies focused on the biosynthesis of benzoxazinoide compounds, the cyclic hydroxamic acids 2,4-dihydroxy-1,4-benzoxazin-3-one and 2,4-dihydroxy-7-methoxy-1,4-benzoxazin-3-one (DIBOA and DIMBOA, respectively), identifying five key responsible genes in maize [58]. In this regard, a mutation in the Bx1 gene in maize demonstrated a clear correlation between gene (Bx1) and metabolite (DIMBOA). Then, a deeper study of the same biosynthetic pathway led to the identification of other genes (named Bx2 through Bx5) encoding cytochrome P450-dependent monooxygenases [58]. Similarly, Song et al. [59] investigated in hydroponic experiments the weed suppressive ability of several rice accessions and banyardgrass (Echinochloa crus-galli) exposed to different nitrogen supplies, identifying two contrasting lines (accession PI312777, highly allelopathic, and accession Lemont, with low suppressive activity). After a subtractive hybridization suppression, to construct a forward SSH-cDNA library of PI312777, the authors sequenced and annotated 35 clones, identifying genes related to allelochemicals. In particular, they reported that in the accession PI312777, the phenylalanine ammonia lyase (PAL) and cytochrome P450 genes strongly increased their transcript abundance at a low N level, suggesting that the higher ability of PI312777 to suppress banyardgrass might be connected to the stronger activation of genes involved in de novo synthesis of allelochemicals [59].
Besides gene expression and transcriptomic analysis, the involvement of PAL-related genes in rice allelopathy was further elucidated using proteomics and bioinformatics approaches. A significant correlation between inhibitory effects of allelopathic rice on weeds and a higher expression of PAL in the phenylpropanoid metabolism was demonstrated using the RNA interference (RNAi) approach to silence this gene in rice, highlighting also a quali-quantitative modulation of microorganisms in the rhizosphere [39,51].
2.2. From Genome to Gene
The first technique to sequence a gene or genome dates to 1977 by Sanger and Coulson [73], who revolutionized research in biology by contributing to a new viewpoint in molecular biology. This method, among others [74], dominated genetic research up to 2005 [75,76], in which a new method based on automated capillary electrophoresis was developed to improve data knowledge of genes and genome sequences [77], generating pivotal information related to genetics, epigenetics, and transcriptomics. This new technology, named next-generation sequencing (NGS), has opened new scenarios in health, environment, and agriculture-related studies due to the highly accessible low-cost and fast high-throughput sequencing technique [78]. In addition, the capacity to obtain large genomic data sets (Giga base), the scalability, the de novo sequencing and resequencing, the discovery of genomic variants, and molecular markers in crops are other features that distinguish NGS from the older technologies. Moreover, NGS technologies can be applied for TF binding site identification and chromatin alteration studies [74].
Concerning the first aspect (from weed to crops), the study by Guo et al. [56] is an important milestone in the elucidation of the allelopathic mechanisms implemented in barnyardgrass. Since barnyardgrass is among the most widespread weeds in the world, the authors shed light on the molecular mechanisms underlying its high allelopathic potential. In particular, using genome assembly and annotation (RNA-seq), the authors identified two gene clusters, involved in DIMBOA and phytoalexin momilactone A biosynthesis, which were activated in response to cocultivation with rice, and detoxification seemed to be the main mechanism conferring an extreme adaptation to the weed [56].
However, the limits of this work could be traced back to the polyploid nature of the species used and the technologies adopted as they do not allow to associate cause–effect in a statistically significant way.
2.3. Plant Breeding in Allelopathy
The main goal of plant breeding is to maintain the quality of life on earth. Improving crops for allelopathy falls within sustainable agriculture, and different strategies, based on genetic variability or transformation, can be adopted to reach this goal. In the first case, natural genetic variability is used to obtain multiple genotypic variants with small phenotypic effects, whereas the genetic transformation forms variants with a significant effect on phenotype. Genetic variability among and within species provides a genetic pool on which to select crop with high allelopathic ability [47], highlighting how improving allelopathy in crops depends on the understanding of the genetic control of these traits. However, as demonstrated by different authors, allelopathic traits follow a normal distribution, thereby outlining the quantitative nature of the traits and their polygenic control [48,81]. Thus, the approaches used to study and understand multiple traits in plants are quantitative trait loci (QTL) mapping and genome-wide association studies (GWAS). QTL mapping is based on statistical analysis that links phenotypes, and in this context, it is represented by allelopathic traits, with genotypes (chromosome regions) [81]. In recent years, genetic studies have been performed only on crops with considerable economic importance, such as wheat and rice (Table 1). A full-bodied study was performed by Olofsdotter et al. [82], who report some examples in rice. However, it is important not to overlook the environment’s effect on these quantitative traits. Indeed, for these studies, fixed segregant populations are needed for QTL analysis, in which near isogenic lines (NILs), recombinant inbred lines (RILs), and doubled haploid lines (DHLs) represent the most used plant material. Initially, with the lack of current technology, segregation ratios and first-generation molecular markers were taken into consideration, as well as euploid, aneuploid, and substitution lines, to investigate the loci of genes controlling the accumulation of DIMBOA on chromosomes [83]. Yet, QTLs associated with allelopathic traits in rice were also identified using restriction fragment length polymorphism (RFLP) markers in the F2 population from a cross between two contrasting varieties for this trait [84], while Wu et al. [47] identified such QTLs studying the allelopathic effect of wheat on ryegrass using the “equal-compartment agar method”.
High-throughput SNP genotyping was recently adopted to identify QTLs associated with the allelopathic traits in rice [45]. For this purpose, 98 F8 RILs were produced by single-seed descent by crossing a cultivar with high allelopathic potential (Sathi) with a nonallelopathic cultivar (Non-an). On chromosome 8, two QTLs, qlTL-8 and qlSL-8, were detected and were responsible for shoot and root length inhibition, explaining 20 and 15% of the phenotypic variation, respectively. Interestingly, between these QTLs, 31 genes were located [45].
2.4. Microorganism in Allelopathy
Microorganisms colonizing the rhizosphere, mycorrhizosphere, and phyllosphere play a pivotal role in plant health and performance through different mechanisms, including allelopathy [86,87]. There is continuous allelopathic crosstalk between plants and the complex microbial communities in which plant roots secrete a variety of molecules able to shape the rhizosphere microbiota, which in turn produce feedback on the plant [88,89,90]
The role of root-exuded coumarins in shaping the root microbiome clearing the rhizosphere from competing microorganisms to give coumarin-resistant microorganisms a competitive advantage has been recently highlighted and represents a good example of how plants affect soil microorganism communities [91]. Subsequently, Stinglis et al. [92] described the molecular basis of the Arabidopsis thaliana–Pseudomonas simiae WCS417 beneficial model system, in which the plant and the probiotic rhizobacteria closely collaborate to induce the root-specific MYB72 TF and the MYB72-controlled β-glucosidase BGLU42 scopoletin-dependent biosynthesis. The excretion of this metabolite selectively inhibits the soil-borne fungal pathogens Fusarium oxysporum and Verticillium dahliae and promotes the growth of rhizobacteria P. simiae WCS417 and Pseudomonas capeferrum WCS358 responsible for the rhizobacteria-induced systemic resistance (ISR). This molecular collaboration led to plant protection and growth enhancement and improved the niche establishment of the microbial partner as well.
The phenolic acids secreted by allelopathic rice into soil induced the gathering of myxobacteria in the rhizosphere. The latter is responsible for the production of a large number of secondary metabolites with allelopathic activity, among which quercetin, a potential allelochemical deriving from the ferulic acid-induced Myxococcus xanthus cultured medium and playing a role in weed germination and growth suppression. In addition, Escudero-Martinez et al. [94] identified the QRMC-3HS genomic region as the major determinant of the composition of barley rhizosphere microbiota communities. Then, performing a root comparative RNA-seq profiling on the barley lines with contrasting alleles at QRMC-3HS, they identified a nucleotide-binding leucine-rich repeat (NLR) gene among the primary candidate genes.
In their turn, microorganisms produce allelochemicals such as phytohormones (e.g., ABA, auxins, ethylene), volatile organic compounds (e.g., ketones, alcohols, alkanes, terpenoids), quorum sensing molecules (e.g., N-acylhomoserine-lactones, AHL), and antibiotics [95], that can promote plant growth [96], resistance to stress [96,97], induce resistance to diseases, antagonize phytopathogens [98], and control weeds [42,99].
Among rhizosphere microbiota, plant growth-promoting rhizobacteria (PGPR) is a group of beneficial microorganisms (fungi and bacteria) that can promote plant growth by regulating phytohormones synthesis/transport and inducing plant systemic resistance and tolerance through VOCs production. The RNA-seq profiling revealed that VOC treatment affected the expression of 123 genes, among which cell wall modification, auxin induction, stress, and defense response-related genes, with a notable downregulation of several stress-related genes. Furthermore, a transcriptome analysis of the growth-promoting effect of VOCs produced by Microbacterium aurantiacum GX14001 on tobacco (Nicotiana benthamiana) revealed that most of the upregulated genes in response to the bacterium VOCs were involved in plant hormone signal transduction, phenylpropyl biosynthesis, plant–pathogen interaction, and flavonoid biosynthesis pathways. The authors suggested that plant hormone signal regulation was the way by which GX14001’s VOCs promoted tobacco growth, a suggestion that was validated by further Arabidopsis mutant experiments [101].
Moreover, Berendsen et al. [102] defined the rhizosphere microorganisms as the “plant secondary genome” reporting the impact of the microbe-derived compounds on plant performances. In addition, Rout et al. [103] exposed an interesting perception of the plant microbial genomes as integrated components of the plant genome and highlighted the importance of considering the plant microbiota (all microorganisms) as a plant microbiome (all microbial genomes) that constantly dialogues with the host genes [104]. In addition to the allelochemical substances, a more elaborated communication process, a kind of “molecular allelopathy”, was recently discovered. The “cross-kingdom RNAi” phenomenon is described as a bi-directional communication channel organized by the plant and its associated rhizospheric microorganisms through extracellular vesicles (EVs) carrying miRNAs to induce gene silencing (Figure 1) [105].
This entry is adapted from the peer-reviewed paper 10.3390/agronomy12092043
This entry is offline, you can click here to edit this entry!