Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Polymer Science
In the last few decades, self-healing polymeric materials have been widely investigated because they can heal the damages spontaneously and thereby prolong their service lifetime. Many ingenious synthetic procedures have been developed for fabricating self-healing polymers with high performance.
- fast self-healing
- elastomers
- healing mechanism
1. Introduction
Polymeric materials have become one of the most widely used structural and functional materials due to their light weight, high stretchability, good processability, and multi-functionality [1]. However, compared with metals and ceramics, polymeric materials exhibit a poor mechanical strength, which makes them vulnerable to damage [2]. The creation of structural injuries will unambiguously lead to a significant decline in the mechanical properties and functions, or even to the failure of the related products for further service, which severely limited their practical applications [3,4]. Endowing the polymeric materials with a self-healing capability will certainly improve the stability and safety of them in practical applications and prolong their lifetime. Therefore, the design of self-healing polymeric materials with multiple functions has attracted increasing interest [5,6,7,8,9,10,11,12,13].
Generally, the self-healing polymers can be divided into two categories depending on their self-healing natures. The first category is named as extrinsic system, which is fabricated by embedding microcapsules with reactive fluids or agents within the polymer matrix [14]. The self-healing is realized in a way that the reactive fluids or agents flow out upon injury of the microcapsules caused by damage, which subsequently trigger the in-situ chemical reactions to heal the damaged parts. It is clear that the self-healing ability will lose after completely exhausting of the reactive fluids or agents. For the other category, as illustrated in Figure 1, the materials themselves contain some kinds of reversible dynamic covalent bonds, such as the Diels–Alder reaction and disulfide bond [15,16], or/and various kinds of physical interactions, such as metal-ligand, hydrogen bond, and electrostatic interactions [17,18,19,20,21,22], which makes the self-healing to be an intrinsic nature of the materials. This kind of material is referred to as an intrinsic system and can heal the damages repeatedly. Moreover, the self-healing polymers can be categorized into autonomic and non-autonomic ones depending upon the initiating condition or pathway of the self-healing. It is not hard to understand literally that the autonomic one can repair damages spontaneously through chemical reactions or recovery of the intermolecular interactions without the need of any kind of external stimuli. The other one, the repair of damaged polymers can only be achieved through some kinds of external stimuli such as the heat and light.
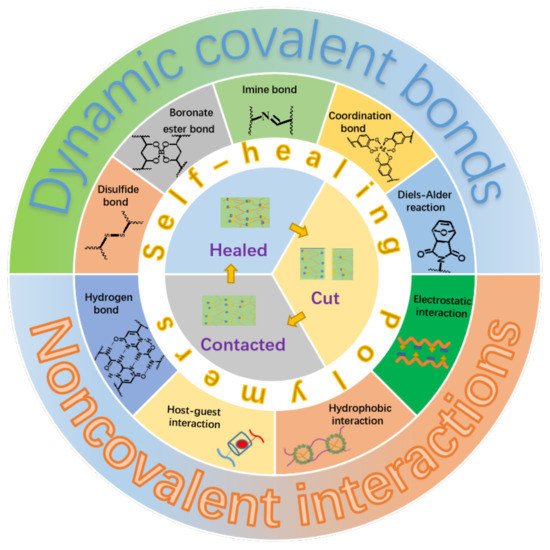
Figure 1. Illustration of self-healing elastomers based on different dynamic covalent bonds or/and non-covalent interactions.
Even though the concept of self-healing polymer was first proposed in 1950s [23], the development of it was insufficient until the 21st century. The polymeric materials with self-healing ability have then attracted much attention since 2000, and great progress has been achieved during the last 20 years. Its development can be divided into realization, blossoming, strength, mild-condition, and fast-healing stages, according to the chronological advancement, namely the perspective of their research priorities at different periods. The first stage focuses mainly on the realization of polymers with self-healing ability, while performances related to other aspects, e.g., tensile strength and functionality, are of less concern. Along with the appearance of self-healing polymer, the investigation of it has blossomed quickly [24]. In the stage referred here to as “strength”, attentions were paid on the mechanical properties of self-healing polymers in order to satisfy the demand of practical applications. At this time, a great number of self-healing polymers with high tensile strength have been reported [25,26,27,28]. While it is recognized that the self-healing of materials with high tensile strength takes place generally at high temperature, sometimes higher than 100 °C, which limited their application, the development of self-healing polymers moves to the fourth stage referred to as “mild-condition” in this paper. Among many ingenious designs such as mechano-responsive strategy [29], novel dual physical cross-linked network [30], molecular engineering of hard domains [31], and mechano-responsive hydrogen-bonding array [32], a milestone work in this stage achieved by Sun et al. [33] shows that the polymeric composite with a high tensile strength of 81 MPa can achieve 99% healing efficiency with the assistance of immersing in 45 °C water. Even ultra-robust (53 MPa) materials with high healing efficiency (80–100%) at room temperature has been reported by Zhang et al. [34].
It should be noted that with the rapid progress of bio-integrated electronics, such as, electronic skins, implantable electronics, and wearable sensors, the flexible electronics based on elastomers have drawn increasing interest [35,36,37,38]. However, the healing time of self-healing elastomers is usually consuming (sometimes lasting for few hours), which results in a long-term failure of the electronics [39]. Consequently, the development of self-healing elastomers with rapid healing ability is highly desired. This makes the design of fast self-healing elastomers, ideally with real-time healing ability at ambient environment, becomes a challenge in this field.
2. Healing Process of Intrinsic Self-Healing Polymers
Due to the potential technological relevance in various fields of self-healing polymers and the substantial for sustainable development, endowing the polymers with self-healing capability and exploring the fundamental mechanism underlying the healing process have attracted increasing interest [40,41,42,43]. In early 1980s, a theory of crack healing of polymers was first established by Wool et al. [44,45] in which the crack healing process of thermoplastic polymers was divided into five parts, termed surface rearrangement, surface approach, wetting, diffusion, and randomization. In a fresh crack surface, the surface rearrangement is mainly in the forms of topographic evolution or roughness of the surface, molecular weight distribution, and chain-end distributions [46]. Plus, compared to the linear molecules, the star-shape ones can form more time-stable networks. [47] Suitable pressure is applied in “surface approach” process to ensure the two freshly damaged surfaces contacting closely together in order to form an interface and wetting each other prior to the molecular level diffusion. The molecular level diffusion plays a key role in the formation of new interactions at the interfaces, which regulates the healing strength of healed polymers. To understand the diffusion at interfaces, the reptation model was used to investigate the healing at polymer–polymer interfaces, which depicted the decrease of healing rates with increasing molecular weight in a power law dependency and the proportional dependence of healing strength on the average interpenetration distance [48,49]. It should be pointed out that the self-healing process of the intrinsic system based on various dynamic bonds or/and different kinds of physical interactions is much more complex due to the multiple and diverse dynamic interactions in different individual systems [50].
Taking the self-healing polymer based on hydrogen bond as an example, the supramolecular polymers with self-healing capability was first established by Cordier et al. [51], who designed a self-healing supramolecular rubber based on small molecules assembled through hydrogen bonds. The supramolecular rubber could recover completely its mechanical properties after 3 h healing at room temperature. It is well documented that the strength of the associations based on the hydrogen bonds is lower than the one on the covalent bonds. Therefore, a large number of dissociated hydrogen bonds present at the fracture surfaces as broken, which endow the supramolecular rubber with efficient self-healing ability through reforming of the hydrogen bonds. This proposed mechanism of self-healing has been subsequently verified by testing the healing efficiency of the samples after being healed for different waiting times. The fact that the healing efficiency decreases with increment of waiting time reflecting the reduction in number of dissociated groups owing to the formation of new bonds within the broken surface during waiting, and thus the reduction of non-associated groups on the fracture surfaces available for self-healing. The aging-time-dependent healing efficiency has, actually, been observed for the majority of the reported self-healing polymers based on hydrogen bond [52,53,54]. Moreover, apart from hydrogen bond based self-healing polymer, self-healing polymers assembled through metal-ligands or ionic interactions also exhibit the similar aging sensitivity [55,56,57]. It was ascribed to the water molecules in air which take part in a series of irreversible processes by coordination with metal ions or ion clusters destroying the noncovalent cross-linkers. It is clear that the evolution of dynamic bonds on the fracture surface plays an important role in surface rearrangement stage for the self-healing polymers based on dynamic bonds.
In the self-healing process, when the two fractured surfaces were brought into contact, the wetting and the reversible bonds re-association took place at the interfaces prior to the molecule-level diffusion [46]. Interestingly, in the systems without dynamic bonds exchange, wetting can also impact the subsequent self-healing stage, and even determine the kinetics of healing process [58]. In order to obtain good wetting, almost all of the self-healing polymers based on hydrogen bonds were applied a certain pressure on the damaged interfaces during the self-healing process. [29,30,31,32]. Moreover, except for the physical interactions based self-healing polymers, dynamic covalent bonds, e.g., disulfide bond and boron-based bond, based self-healing polymers also needs external pressure during the self-healing process to achieve satisfactory self-healing performance (see Table 1) [59,60,61,62,63,64]. A recent study based on experimental and theoretical simulations suggests that the compressive force on the healing interfaces maybe attenuate the activation energy barrier of dynamic bond exchange that can further promote the healing in the manner of accelerating dynamic exchange [65].
Table 1. External pressure assistant self-healing polymers based on dynamic covalent bonds.
We recall that the molecular-level diffusion is essential for self-healing, and the damaged sample can only recover its initial mechanical properties when the healed interfaces exhibit indistinguishable structure from the pristine sample [66]. Quantitative study on the evolution of the healed interfaces, especially the interfacial diffusion in microscopic scale during the healing process is, however, difficult. Therefore, the macroscopic experiments, e.g., disappearance of scratches, restoration ratio of tensile strength and strain are used to evaluate the healing performance of self-healing polymers. Moreover, the internal reflection infrared imaging [67] and laser speckle imaging [68] have been used to explore the microscopic healing process, even though their resolutions are not high enough. Recently, Schrettl et al. [69] reported a unique stratagem to monitor the interfacial diffusion in healing process with a high resolution of a few nanometers by energy-dispersive X-ray (EDX) spectrum imaging using scanning transmission electron microscopy. Heterogeneous interfaces as illustrated in Figure 2a were constructed to investigate the interfacial diffusion of the healing process by monitoring the diffusion of Eu3+ and Tb3+ (Figure 2b). They pointed out that a mixed interphase of more than 100 nm and less than 175 nm was required to achieve complete recovery of the mechanical properties. This study provides a direct proof for the self-healing process that molecular-level diffusion is essential to heal the cracks. Furthermore, it establishes a framework for further investigation of the healing process in intrinsic self-healing system based on the dynamic bonds.
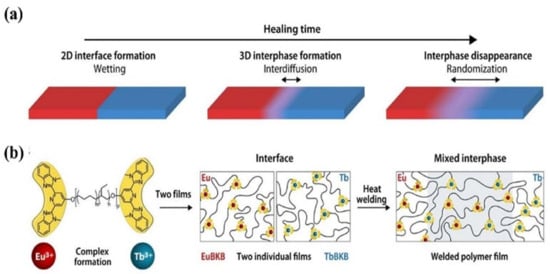
Figure 2. The healing process in polymers. (a) The final stages of the healing process in polymers involve wetting, interdiffusion with re-entanglement, and randomization. (b) To investigate the healing process on a length scale of a few nanometers, metallosupramolecular polymers (MSPs) assembled from telechelic PEB with terminal Mebip ligands (Mn = 3800 g mol−1; m ≈ 0.32, n ≈ 0.68, p ≈ 55) and either Eu(ClO4)3 or Tb(ClO4)3 were studied. The two metallosupramolecular polymers display similar properties, but the different ion types can be monitored in a spatially resolved manner. Reproduced with permission [69]. Copyright 2021, American Association for the Advancement of Science.
This entry is adapted from the peer-reviewed paper 10.3390/ma15175993
This entry is offline, you can click here to edit this entry!