1. Introduction
Cells are constantly exposed to a huge number of intra-and extra-cellular signals which requires a proper cell response for adaptation and homeostasis control [
1]. Among the different sensors and integrators for such varying signals, the family of G-protein coupled receptors (GPCRs) arise as one of the most important transducers [
2,
3]. With over 800 GPCRs encoded in the human genome, this family of transmembrane receptors can bind a plethora of stimuli which include hormones, metabolites, and inflammatory mediators, influencing a diverse network of signaling pathways [
4].
Apart from the classical chemical inputs, recent studies have begun to unravel the potential of mechanical and architectural properties of the environment as a new and alternative way to dynamically modulate cellular homeostasis [
5]. Extracellular Matrix (ECM) is considered as an intricate meshwork of proteins and carbohydrates organized in a specific manner which acts not only as a reservoir of growth factor and bioactive molecules but also as a highly dynamic entity which provide mechanical rigidity and structural support to the cells [
6].
All GPCRs contain seven transmembrane domains embedded within the cell membrane with several intracellular domains that trigger guanine nucleotide binding proteins downstream signaling pathways and, interestingly, extracellular domains in GPCRs have been reported as potentially critical elements in the interaction with components of the ECM and as a force sensing mechanism [
7,
8]. This opens the questions about the potential of GPCRs as linkers in the integration of all these signals (both chemo- and mechanical stimuli) raising the possibility that specific GPCRs and its downstream effectors can mediate the crosstalk between both types of triggers during the control of homeostatic feedback loops. Indeed, the ECM has the potential to significantly impact virtually on every physiological cellular mechanism [
9,
10]. Excessive deposition and increased stiffness of ECM has been directly linked with the progression of many different pathologies and has the potential to regulate cellular metabolism [
11].
An important downstream process in the crossroad between chemo- and mechanosensing regulatory responses is autophagy [
12]. The catabolic activity of autophagy is a fundamental cellular process that eliminates molecules and subcellular elements (including proteins, lipids, nucleic acids and even organelles) through a lysosomal-mediated degradative pathway for providing energy sources for ATP production or building blocks for protein synthesis [
12]. Activated by different types of stress including those related with DNA damage, hypoxia, oxidative stress, inflammation, and food deprivation and by different challenges arising from mechanical (stretching, shear stress or hypotonic shock), autophagy can have both beneficial and deleterious effects [
13,
14,
15]. Indeed, vascular and heart diseases, infectious diseases, neurodegenerative pathologies, and cancer have all been related to autophagy dysfunctions. Thus, autophagy represents a double-edge sword and for this reason the possibility to regulate autophagy in a time- and local-dependent manner represents a novel and valid therapeutic approach to control most of these disorders.
2. Autophagy between Nutrient, Mechanical and Oxidative Stress: An Emerging Role of Gαq
Autophagy is a highly conserved mechanism for cellular degradation in which cytosolic waste, protein aggregates and organelles are sequestered into a double-membrane vesicle (autophagosome) and delivered into lysosomes for breakdown [
120]. Autophagy is orchestrated by sequential steps tightly control machinery in which ATG and associated proteins regulate the formation and maturation of autophagosomes into autolysosomes [
121]. Under pathophysiological conditions, damaged or excessive accumulation of organelles such as endoplasmic reticulum, ribosomes, mitochondria, lipid droplets and peroxisomes can be degraded through mechanisms mediated by a collection of specific autophagy-related proteins [
13,
120].
Typically, autophagy stimulation depends on the mTOR system modulation [
122]. It is initiated by the ULK1 complex (Unc-51-like-autophagy-activating kinase) which can receive input from cellular energy balance, and the availability of nutrients from mTORC1 (Rapamycin Complex1) and AMPK signaling networks (AMP-activated protein kinase). Canonical initiation of autophagy entails those metabolic stresses (chemical stimuli) such as nutrient deprivation. This causes mTORC1 dissociation from ULK1 which becomes active and binds to ATG13 and FIP200 triggering autophagosome formation [
123,
124]. In addition, mechanical stresses are also involved in autophagic flux control. Although it is unclear whether mechanical stresses may play a direct role in ULK1 activation, it has been reported that the mechanosensitive mTORC2 complex can indirectly induce ULK1 activation via inactivation of mTORC1 repressor function, through a FAK (focal adhesion kinase)-dependent mechanisms [
125,
126,
127].
The extracellular matrix (ECM) constitutes a dynamic and plastic network of biophysical and biochemical factors that maintains tissue homeostasis. Changes in ECM composition, elasticity, and structure have also been reported to impact on autophagic flux raising the potential of matrix biology modulation as a critical controller of this process [
9]. Apart from the interaction with physicochemical environmental imposed by the ECM, cells are also subject, and they have to respond to a great variety of mechanical forces due to other external forces, such shear stresses of fluid pressure (e.g., blood vessels), lateral stretches and compression (such in the case of muscles). Overall, this plethora of short- and large- scale forces elicit an adaptive cellular response in which autophagy seems to be a critical player.
In this adaptive response, cells can sense extracellular mechanical cues in different ways. This includes cellular adhesion complexes with ECM and/or cells, mechanosensors such as proteoglycans localized at the cell surface or mechanically activated ion channels (e.g., Piezo) [
128,
129] even plasma membrane-associated structures such cilium, caveolae, and clathrin-coated pits [
115,
130,
131]. Moreover, multiple intracellular organelles, including autophagosomes, can also sense mechanical forces [
132,
133,
134].
Mechanical cues imposed by forces or microenvironmental cues may affect the autophagic process through specific crosstalk with autophagy regulatory proteins (such as mTORC system, or AMPK pathway) or via mechanical regulation of cytoskeletal elements or phospholipid membranes, which can be crucial in the autophagic process [
135,
136,
137]. Interestingly, a direct link between cell attachment to ECM and autophagy has also been reported [
138]. Loss of cell attachment with the ECM usually results in programmed cell death via anoikis. In some cases, ECM-cell detachment can rapidly activate autophagy allowing for survival and re-attachment to the substrate [
139]. However, it remains elusive the mechanism controlling this process with integrin-mediated adhesions emerging as a critical element [
140]. Furthermore, ECM and integrin-mediated adhesion may trigger autophagy via FAK and ILK (integrin linked kinase) being relevant in different processes such as for immunosurveillance [
141].
Additionally, matrix constituents have been shown to regulate autophagy in both directions, promoting or inhibiting this process, depending on matrix stiffness but also on its specific composition [
142]. Indeed, recent studies have demonstrated an active and dynamic signaling role of specific extracellular matrix components on autophagic regulation which can act in both positive (activators) or negative (inhibitors) ways. Among them, decorin, collagen VI, kringle 5, endorepellin and endostatin function as activators and pro-autophagic matrix constituents that engage a diverse array of cell surface receptor for autophagic initiation [
143,
144,
145]. In contrast, laminin α2 acts as an inhibitor. Thus, absence of laminin α2 permits excessive autophagy [
146]. Interestingly, the action of all these components seems to be independent of the predominant nutrient concentrations. This unique class of matrix molecules can function as an alternative mechanism to the classical nutrient deprivation mechanism to safeguard cellular homeostatic balance through autophagic control and providing a new mechanism through which GPCRs could also be participating in the regulation of autophagy.
As we mention in a previous section, GPCRs are directly linked with mechano-transduction mechanisms (see
Table 1). Supporting this idea, it has been demonstrated that mechanical perturbations can modulate GPCR conformational transitions [
36]. Moreover, the response to shear stress can be directly modulated through the Gαq-coupled GPCR, GPR68 [
35,
41]. Interestingly, PKCζ, a protein that we have described as new interactor of Gαq [
147], can be regulated by shear stress and activated by disturbed flow in atheroprone areas [
148]. Further evidence on this mechanical stimulation comes from studies on adhesion GPCRs which display a long extracellular N-terminus with adhesive properties to the extracellular matrix or N-glycans modifications. These glycan chains have been reported to be able to be activated in the context of mechanical traction forces [
38,
149]. Further investigations are required to really address how these forces can structurally activate GPCRs in different contexts.
As a sequential process involving membrane remodeling events, autophagy is mechanically linked to cytoskeletal dynamics that lead to mechanical deformation and transport. Actin filaments and fibers and microtubule network can act as critical modulators in the control of organelle dynamics and autophagy control [
150].
Although the most classical autophagy process relies on the delivery of cytoplasmic material to lysosomes via the double-membraned autophagosome, another form of autophagy, known as chaperone-mediated autophagy (CMA), occurs in the lysosome directly [
151]. Thus, lysosomes can be considered critical hubs in the modulation of autophagic control. Lysosomes constitute a highly dynamic organelle which display important changes, including acidification and enhanced enzymatic activity. Furthermore, this organelle can move from the perinuclear localization to the cell periphery with important implications in cell metabolic control [
152].
An emerging aspect to be considered is how lysosomal position can directly modulate its function. Lysosomes are transported bidirectionally through the microtubule network by dynein and kinesin motors, with microtubule motors such as dynein modulating the movement of lysosomes from the periphery towards a perinuclear location, while kinesins promote the scattering of lysosomes through the cytoplasm [
153,
154]. Recent evidence suggest that the distribution of lysosomes can be controlled by stimulation with different inputs. Under cell starvation, autophagosomes and lysosomes move toward the center of the cell facilitating the fusion of both compartments and the degradative capacity [
155].
Several protein complexes have been implicated in the regulation of lysosomal positioning. One important regulator is the Rab7, a small GTPase involved in the coordination of appropriate coordination and homeostatic control of late endosomes and lysosomes [
156,
157]. Recently, WDR91 a Rab7 effector has been reported to be essential for lysosomal function [
158]. In addition, the transcription factors TFEB and TFE3 are essential to promote the expression of multiple lysosomal genes. They play critical roles in the modulation of lysosomal biogenesis and distribution through the control of the lysosomal transmembrane protein TMEM55B (transmembrane protein 55B) expression [
159]. TFEB acts as a link between autophagic process and lysosome biology [
160], interacting with mTORC1 complex but not with mTORC2 controlling the lysosomal localization and function of mTORC1 [
161].
Our recent studies strongly suggest that Gαq is a critical autophagy regulator raising the potential to control the shift between mTORC1-mTORC2 switch through lysosome control. We have recently reported a higher number of autolysosomes in cell lacking Gαq/11 compared to the wild type of counterpart which is directly linked with the involvement of this protein in the modulation of autophagy [
21]. Immunofluorescence with LAMP1, a lysosomal-endolysosomal compartment marker, revealed that Gαq KO cells showed a predominant perinuclear lysosomal distribution in basal conditions, a phenotype that was mimicked by upon starving conditions. These results are consistent with a not-yet described role of Gαq in the modulation of lysosome dynamic regulation.
Furthermore, although many components of the autophagic machinery and autophagy receptors which are involved in the regulation of the process are being subjected to lysosomal degradation, in the case of Gαq, its presence in lysosomes does not alter its protein expression levels neither in basal nor in nutrient stress conditions which reinforce a critical role of this protein in the modulation of the autophagic control process.
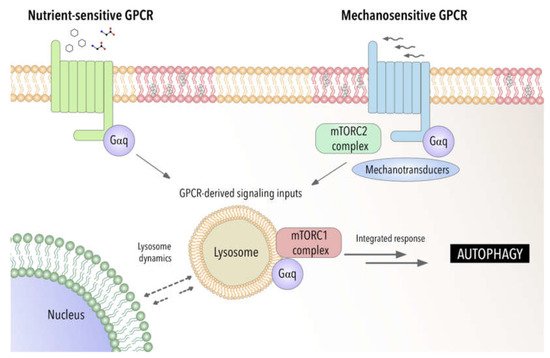
Recent studies have demonstrated by using a biosensor the presence of GTP-loaded Gαq/11 at endosomes [
162]. This provides a powerful tool to be applied to other cellular organelles such lysosomal compartment to fully address the specific function of GPCR- Gαq signaling at these organelles. Furthermore, this raises the possibility that GPCR-Gαq signaling may act as a modulator of autophagy by acting as a switch between chemical and mechanical cellular responses (see
Figure 1).
Figure 1. Gαq as a potential integrator of chemical and mechanical signals modulating autophagic process. Involvement of Gαq interactome-autophagy control in pathophysiological settings.
3. Autophagy in Disease for Good and for Bad: Gαq Involvement
Organisms have to constantly adapt to external stimuli and changes in their intracellular environment. Organs, tissues and cells have to face both chemical (e.g., Ca
2+, amino acids, cytokines, chemokines and hormones) as well as physical challenges. Among the cytoplasmic responses to mechanical forces, recent studies have uncovered the role of autophagy in the translation of mechanical forces into biological responses [
126,
127,
192,
193]. Given the importance of autophagy regulation and dynamics of lysosomal system to ensure cellular fitness, it is not surprising that autophagy disruption can contribute to the development of several diseases such as metabolic disorders, cardiovascular or cancer diseases. Although the involvement of autophagy in these major diseases has been well studied (reviewed in [
194], over the years autophagy regulation has grown in complexity and their consequences are less predictable. The importance of Gαq and Gαq-coupled GPCRs in all these contexts, together with its recently described importance in autophagy, strongly suggest that alteration in Gαq modulation signaling pathways can contribute to all these pathological situations. In this part of the review, we will focus on how autophagy may be involved in different pathologies, emphasizing as far as possible the influence of mechanical inputs.
Various metabolic disorders have shown functional defects in autophagy [
195,
196]. Since the lysosomal disposal of intracellular macromolecules leads to their breakdown into important metabolic intermediates, including amino acids, glucose, nucleotides, and free fatty acids (FAs), autophagy plays an important role in the response to energetic stresses, at both the tissue-specific and systemic levels [
197]. Many studies have emphasized the importance of autophagy in conditions such as obesity, insulin resistance and diabetes that are characterized by metabolic alterations and intracellular stresses that have in common the accumulation of damaged cellular components. Silencing of ATG system promotes obesity and induces metabolic alterations [
198]. Interestingly autophagy genes are differentially expressed and activated in a tissue and stage-specific manner. In general, nutrient limitation and different stress situations favor autophagy as a mechanism of cytoprotecting, reducing cellular death and limiting inflammatory response. Upon autophagy inhibition alteration of adipocyte differentiation, lipid metabolism and storing of lipids is drastically altered [
199,
200,
201] For example, in obesity, autophagy is suppressed due to an increased in mTOR activity. Moreover, in patients with diabetes, changes in oxidative stress and autophagy have been reported [
202]. Therefore, the enhancement of autophagy activity has been suggested as a novel therapeutic approach against organ failure associated to metabolic disorders.
In general, GPCRs regulate virtually all metabolic processes including glucose and energy homeostasis, particularly diabetes and obesity-related diseases. Several endogenous ligands such as free fatty acids and their receptors have been extensively studied in insulin secretion regulation, and glucose metabolism [
203]. A growing number of GPCR are being identified as sensors of circulating of local concentrators of energy substrates or metabolic intermediates. Examples of these receptors include the amino-acid responsive receptors GPRC6A taste receptors type 1 members 1 and 3 (T1R1/T1R3), the calcium sensing receptor (CaSR) long chain fatty acid receptors GPR120 and GPR40, short fatty acid receptors GPR41 and GPR43 or hydroxy carboxylic acid receptors [
204,
205,
206]. These GPCR nutrient receptors act via different G proteins including Gαq/11 and might be able to modulate the canonical metabolic regulators AMPK and mTORC1 [
19]. In this sense, Gαq-coupled T1R1/T1R3 act as a direct sensor of the fed state and amino acids availability, leading to the activation of mTORC1 [
207]. Nutrient and homeostasis fluctuations may also indicate the release of classical hormones and neurotransmitters that activated GPCRs, along a systemic regulation of autophagy. In this sense, β-adrenergic receptors activation has been related with autophagic flux favoring lipolysis [
208], and hyperglycemia induces autophagy in pancreatic β cells through P
2Y purinergic receptors [
209]. In addition, drugs that target metabolic tissues have emerged as attractive diabetes therapeutic targets as well. The p62-mTORC1-autophagy axis has been described to regulate adipogenesis and energy control in a complex manner [
210]. The potential and reported connections of Gαq signaling that we have described with this axis [
21] may provide new insights in the mechanisms underlying these metabolic alterations. Recent studies have further confirmed the relevance of Gαq signaling for driving metabolic reprogramming in uveal melanoma [
211] and in the regulation of glucose and lipid homeostasis [
212] reinforcing a critical role of GPCR-Gαq system in metabolic diseases. Further investigation will be required to define the mechanisms involved.
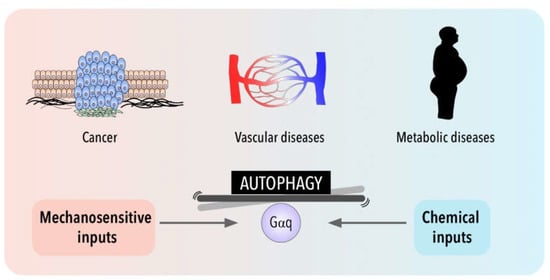
Interestingly, cell metabolism is sensitive also to the physical cell microenvironment [
213]. Although cell metabolism has recently emerged as one of the processes regulated by mechanical cues, the link between cell mechanics and metabolism is still poorly understood when compared with other pathologies such as cardiovascular diseases or cancer (
Figure 2). Thus, in addition to metabolic intermediates, autophagy can influence metabolic reprogramming in epithelial cells through the involvement of mechanical forces such as shear stress [
214,
215] and, mechanical stretching/tension exerted by exercise has been shown to induce also autophagy in peripheral tissues (liver, pancreas and adipose tissue) [
216].
Figure 2. Gαq signaling and autophagy at the crossroads of a balance between mechanical and chemical cues and their impact in cancer, metabolic and cardiovascular pathologies.
The involvement of shear stress and mechanical forces in endothelial function has been well established and although, there is an increasing interest in the role of autophagic flux in vessel wall biology, the mechanosensors upstream of autophagy induction in endothelial cells are not well known [
217]. Emerging evidence links alterations in autophagic flux with disease processes that include atherosclerosis, pulmonary hypertension and cardiovascular diseases [
218]. Interestingly, a very recent study proposes a protective role of CMA (Chaperon Mediated Autophagy) against atherosclerosis [
219]. Loss of autophagy may be a central mechanism through which risk factors elicit endothelial dysfunction. The role of autophagy in vascular development and in sprouting has been associated with a defective autophagy in mice lacking endothelial specific-TFEB factor [
220]. The autophagic state of endothelial cells is also critical for vascular permeability [
221]. Additionally, endothelial cells require autophagy to regulate tight junction proteins and maintain endothelial barrier integrity during inflammation [
222]. Moreover, it has been reported that autophagy may be involved in the regulation of nitric oxide bioavailability, a crucial molecule that maintains vascular homeostasis in endothelial cells. Concomitant with a reduction in NO, loss of autophagy promotes and increase in endothelial ROS and inflammatory cytokine production [
223,
224]. Shear-stress-dependent autophagy is also important for NO production [
224,
225]. Indeed, in zones of low shear stress that are prone to develop atherosclerotic plaques, the impairment of autophagic flux induces endothelial NO synthase (eNOS) uncoupling, resulting in the production of superoxide instead of NO. Restoration of the autophagic flux favors the production of NO by endothelial NO synthase [
226]. Interestingly, Gαq has been described as an important sensor of shear stress in endothelium [
33,
227]. Recent studies have demonstrated that changes in the type of flow can activate the same initial mechanosensing pathway involving Piezo1- and Gαq/11-mediated signaling with different atheroprotective response depending on the activation of α5 integrin, which is activated only by disturbed flow, but not by sustained laminar flow [
228].
Regarding cardiovascular context, autophagy preserves cardiac structure and function under baseline conditions and is activated during stress, contributing to limit damage and preserve cardiac functionality during ischemia [
229]. Cardiac cells are also subjected to tension. Several pathophysiological conditions, lead to an increase in cardiac workload and mechanical forces that are usually associated with pathological cardiac hypertrophy [
230]. Mechanical forces can induce autophagy in cardiac cells being protective or detrimental depending on the context [
217]. Indeed, during ischemia, autophagy has a protective effect on cardiomyocytes [
231,
232,
233], while inhibition of autophagy improves cardiac function after reperfusion in an ischemia–reperfusion mouse model [
232]. Much evidence places Gαq at the center of hypertrophic pathways in the heart (see [
234], for more details). Indeed, Gαq signaling is both necessary and sufficient for the development of cardiac hypertrophy. The development of a cardiac hypertrophy phenotype has been correlated with a higher risk of heart failure [
186]. Interestingly, it has become increasingly clear that both events are tied to the activation/presence of Gαq that can promote cardiomyocyte apoptosis and heart failure [
235] by affecting vascular permeability and hypertension. Indeed, Gαq inhibition using specific drugs have been proposed to have anti-hypertensive role [
236]. We have also described, in previous studies, a role of Gαq/PKCζ signaling axis in the development of cardiac hypertrophy in response to angiotensin II through a novel binding region on Gαq. Given the involvement of Gαq in cardiovascular function and in the process of autophagy, the potential participation of novel mechanisms downstream Gαq directly linked with autophagic flux in cardiovascular system is a relevant open question.
Autophagy is also recognized as a critical player in a context-dependent manner in cancer. Although it is well accepted that autophagy is important in many diseases, as described above, practically all clinical studies that involve autophagy manipulation are focused on cancer therapy. Autophagy networks are related to multiple aspects of cancer and may play a dual role with tumor-suppressive and tumor promoting functions depending on tumor cell type and stage [
237]. Both inhibition of autophagy and its overstimulation are strategies tested in cancer, with the use of different drugs such as hydroxychloroquine, 3-methyl-adenina and everolimus as currently new strategies to be employed in clinics in combination with other chemotherapeutic treatments [
238]. However, the high toxicity and adverse effects of these treatments urge a further understanding of the specific mechanisms by which autophagy modulates the different tumor progression steps.
Deficiency of autophagic genes has been found in various cancers. Impaired autophagy can promote tumorigenic environment through ROS dysregulation and inflammation processes [
163,
239,
240]. On the other hand, at advanced cancer stages, increased autophagy can sustain tumor cell growth in nutrient-deficient, hypoxic tumor microenvironment and resistance to anoikis [
241]. Upregulation confers chemoresistance and promotes the maintenance and survival of stem cell cancer status. Furthermore, autophagy inhibition can favor tumor cell invasiveness through the induction of de-differentiation mechanism. Thus, it seems that in premalignant lesions, enhanced autophagy might be beneficial preventing cancer, but in advance cancers most therapeutic strategies are focused on inhibiting autophagy [
242]. Adding another layer of complexity, the novel Gαq role in modulating autophagy suggests that the balance between these processes characteristic of tumor growth might be altered in different cancer settings.
Furthermore, evidence identifies tumor microenvironment as a central driver of tumorigenesis in cancer [
243]. Interestingly, cancer cells can also experience shear stress that can induce autophagy in different tumor cell lines [
244,
245,
246,
247]. Interstitial flow can promote the distribution of tumor-derived cells in primary tumor, while circulating tumor cells are also subjected to the shear stress from body fluids (blood, lymph and interstitial fluid) during metastasis [
248,
249]. It has been suggested that shear stress-induced autophagy can play an important role in controlling important cell responses from the regulation of cell size and metabolism to inflammation and cell death [
217]. Moreover, the activation of tumor stromal fibroblasts to a state commonly known as cancer associated fibroblasts (CAFs) is critical. CAFs impact tumor progression by the modulation of multiple secretion functions of different factors (growth factors and inflammatory signals), by remodeling the extracellular matrix and even reprogramming their metabolism to provide nutrients and survival factors [
250]. Moreover, autophagy can play a key role in CAFs activation [
251]. Recent studies demonstrate that normal fibroblasts can differentiate into CAFs as protective responses to stresses under tumor microenvironment via the p62-Nrf2-pathway [
252]. Furthermore, a molecular mechanism for CAFs activation has shown that tumor secreted lactate downregulates p62 in the stroma blocking AP-1-mediated p62 transcription [
253]. Interestingly, we have described changes in the expression of Gαq affecting some partners such as p62 promoting its downregulation and favoring autophagic flux [
21].
Altered GPCR pathways have increasingly been reported in cancer context and activating mutations in Gαq has been identified in approximately 80% of uveal melanomas. Considering Gαq as a component of the nutrient-sensing machinery, able to link nutrient availability with the activation of mTORC1 through its interaction with p62 [
21] strongly reinforce its potential contribution in the modulation of both the tumor and its microenvironment, during tumor progression.
This entry is adapted from the peer-reviewed paper 10.3390/antiox11081599