Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
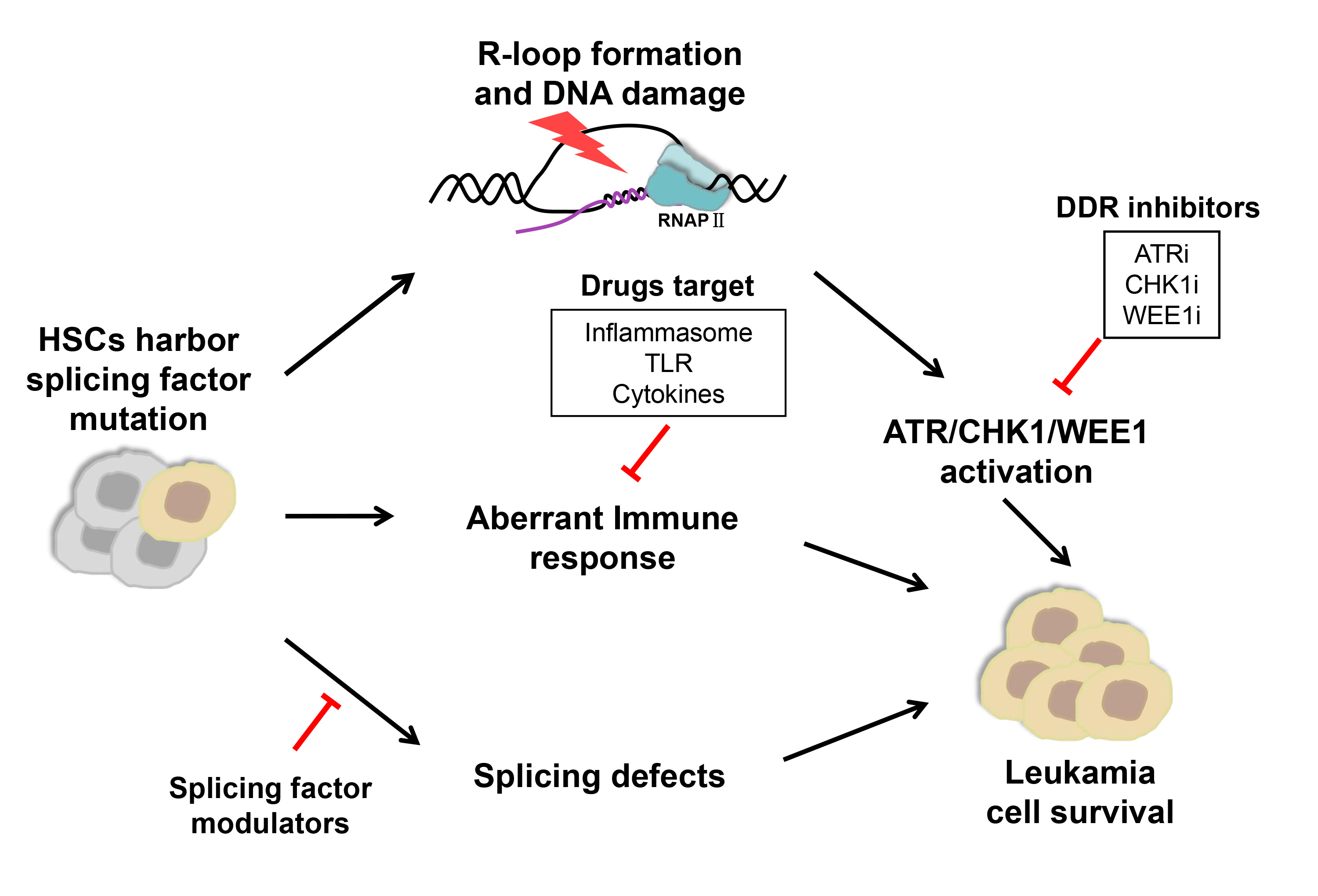
Splicing factors are frequently mutated in myelodysplastic syndromes (MDS) and acute myeloid leukemia (AML). These mutations are presumed to contribute to oncogenic transformation, but the underlying mechanisms remain incompletely understood. While no specific treatment option is available for MDS/AML patients with spliceosome mutations, novel targeting strategies are actively explored, leading to clinical trials of small molecule inhibitors that target the spliceosome, DNA damage response pathway, and immune response pathway.
- myelodysplastic syndromes
- acute myeloid leukemia
- spliceosome
1. Introduction
Splicing factors are frequently mutated in a variety of hematologic malignancies [1,2,3], including myelodysplastic syndromes (MDS) that may progress to acute myeloid leukemia (AML) [4,5,6]. Among all splicing factor mutations, major heterozygous point mutations in Splicing factor 3B subunit 1 (SF3B1), U2 small nuclear RNA auxiliary factor 1 (U2AF1), Serine and arginine rich splicing factor 2 (SRSF2), and RNA binding motif and serine/arginine rich 2 (ZRSR2) have been demonstrated as driver mutations to promote disease progression [2,7]. RNA sequencing studies further revealed that all of these mutations altered spliceosome functions, resulting in numerous sequence-specific mis-splicing events [8,9,10,11]. Interestingly, mutations in different splicing factors are mutually exclusive and only heterozygous mutations were discovered in patients [12]. These observations suggest that mutations in different splicing factors may have convergent adverse effects beyond cell tolerance and mutant cells rely on the canonical functions of the wild-type allele for survival [2]. Indeed, recent studies using CRISPR/Cas9 to selectively knock out the wild-type allele of the splicing factor genes confirmed the dependency of mutation-carrying cells on the wild-type allele [12,13].
Mutations in SF3B1, U2AF1, and SRSF2 have also been shown to promote aberrant R-loop formation, which triggers replication stress, followed by DNA-damage responses, specifically the activation of Ataxia telangectasia and Rad3 related (ATR)-CHK1 [14,15,16]. These mutations may elicit innate immune pathways as well, leading to chronic activation of the inflammasome and inflammatory cytokine production [17,18]. All these findings of diverse disease mechanisms have raised a broad interest in developing new therapeutical strategies to modulate splicing regulatory activity, block specific DNA damage repair pathways and target immune response and inflammatory pathways.
2. Direct Targeting of the Spliceosome with Splicing Factor Modulators
As the splicing defect is a direct consequence of mutations in splicing factors, predominant interests have been given to search for key downstream targets of mis-spliced genes in MDS/AML patients. Moreover, as MDS clones harboring splicing factor mutations may be preferentially sensitive to inhibition of the RNA splicing process compared to the unmutated stem/progenitor compartment [30], several drugs targeting the splicing machinery are currently evaluated in clinical trials. In particular, a variety of natural products and their derivatives that bind to the SF3B complex and inhibit pre-mRNA splicing at an early step of spliceosome assembly were discovered, including pladienolides, E7107, FR901464, spliceostatin A, herboxidiene, and sudemycin D6 [87,88,89,90,91]. Extensive efforts have been made in the last few years to examine if these small chemicals have antitumor activity in MDS/AML patients [87,88].
Pladienolide is a natural macrolide [87], and E7107 is a semisynthetic derivative of pladienolide. Treatment of isogenic murine myeloid leukemias with E7107 revealed preferential death of leukemic cells bearing mutated SRSF2 [12,96]. However, further clinical trials of E7107 were halted due to dose-limiting toxicity as participants developed vision loss without clinical benefit [109,110]. FR901464 displays strong antitumor activity and inhibits cell cycle progression at G1 and G2/M phases. However, it exhibits strong cytotoxicity at low nanomolar concentrations as well [92]. In a recent report, the synthetic derivative of pladienolide H3B-8800 appears to hold more promise for clinical application. H3B-8800 has exhibited good binding activity to spliceosomal components in a dose-dependent manner, and is safe even with prolonged dosing in myeloid neoplasms [94]. It is orally bioavailable and inhibits tumors with mutations not only in SF3B1, but also in U2AF1 and SRSF2 in xenograft leukemia models [3,93]. A related Phase I clinical trial (NCT02841540) is currently performed in patients with MDS, AML, or CMML [94,95], to determine its safety and potential therapeutic efficacy.
Spliceostatin A is a methyl ketal derivative of FR901464 and inhibits both splicing and nuclear retention of pre-mRNA [88]. It interferes with spliceosome assembly at the step following the binding of U2 snRNP with pre-mRNA [111]. Recently, spliceostatin A has been found to elicit apoptosis of chronic lymphocytic leukemia cells through downregulation of the pro-survival Bcl-2 family member Mcl-1, the high expression of which is correlated with disease progression [112]. Herboxidiene is a microbial product with antitumor activity, which serves as a novel splicing inhibitor that specifically impairs the function of SF3B by binding to SAP155 [90]. Sudemycin D6 is a small molecule that targets SF3B1 and inhibits spliceosome activity as well [91]. Hematopoietic cells expressing U2AF1 (S34F) are sensitive to sudemycin D6 [113], and treatment of U2af1 (S34F) transgenic mice with sudemycin D6 results in attenuated expansion and increased apoptosis of hematopoietic progenitor cells [114].
In addition, Rbm39 modulators bind to Rbm39 and recruit the E3 ubiquitin ligase CUL4-DCAF15 for Rbm39 degradation, leading to the disruption of the SF3B1 complex formation [97]. Protein arginine methyltransferase 5 (PRMT5) and Protein arginine methyltransferase 1 (PRMT1) methylate spliceosome components are required for spliceosome formation. Protein arginine methyltransferase 5/1 inhibitors reduce spliceosome methylation and inhibit splicing, and leukemias with splicing factor mutations are preferentially sensitive to PRMT5 inhibition [30,98,99]. A few PRMT5 small-molecule inhibitors, GSK3326595, JNJ-64619178, and PRT543, are currently in early-stage clinical trials to evaluate the specificity and efficacy in multiple tumors, including MDS and AML (NCT03614728, NCT03573310, NCT03886831), with the hope to benefit patients in the future.
In theory, splicing inhibitors are expected to possess limited selectivity for tumor versus normal cells. Therefore, the side effect is a potential issue for clinical use of spliceosome inhibitors. Toxicity might be alleviated by lowering splicing modulators’ doses at the expense of therapeutic efficacy. In any case, more work is needed to improve treatment expectations.
3. Targeting DDR with Small Molecule Inhibitors
On one hand, mis-repaired DNA damage promotes genomic instability and mutation accumulation, inducing cell transformation and cancer progression. On the other hand, DNA damage checkpoints can induce apoptosis, senescence, or differentiation, leading to depletion of stem cells and, ultimately, bone marrow failure [72,115]. Chronic DNA damage is accompanied by constitutive activation of checkpoint pathways in primary AML cells with complex karyotypes [116]. One interesting hypothesis is that hematopoietic cells with splicing factor mutations suffer cellular stress in both gene expression and replication, resulting in defective self-renewal and differentiation potential. Some mutant cells may undergo specific transformation and adaptation, and survive the stress, yet become selectively dependent on endogenous DNA damage repair activity at MDS/AML stages.
ATR is a serine/threonine-specific protein kinase that is activated in response to persistent single-stranded DNA, leading to activation of the DNA damage checkpoint and cell-cycle arrest. Previous studies show that malignant cells, including AML samples, are particularly sensitive to hypomorphic inhibition of ATR compared to normal tissues [117]. At the molecular level, ATR blockade results in G2/M checkpoint abrogation, DNA damage, and apoptosis in AML cell lines and primary tumor samples [118]. Interestingly, it has been shown that the transcript of ATR is alternatively spliced in U2AF1-mutant AML patient samples and the mutated U2AF1 CD34 cells [31,119]. Consistently, cells with U2AF1 or SRSF2 mutation are more sensitive to ATR inhibition in vitro [15,74]. AZD6738 is an orally active ATR inhibitor and sensitizes p53- or ATM-defective primary CLL cells to chemotherapy and ibrutinib both in vitro and in vivo assays [120]. MDS CD34+ cells with splicing factor mutations (SF3B1, SRSF2, and U2AF1) are hypersensitive to ATR inhibitors (VE-821 and AZD6738) [73]. Encouraged by these pre-clinical results, AZD6738 is currently in the phase I trial in the treatment of MDS or CMML patients with at least one mutation in splicing factors (SF3B1, U2AF1, SRSF2, and ZRSR2), for safety and tolerability test (NCT03770429) [74,100].
While monotherapy studies aim to exploit specific molecular vulnerabilities of tumors, most clinical trials of ATR inhibitors (ATRi) are performed in combination with other drugs. The combination of ATRi and antimetabolite chemotherapy has been studied in the context of acute myeloid leukemia (AML); ATR inhibition by VE-822 treatment can enhance hydroxyurea- and gemcitabine-induced growth inhibition through S-phase cell cycle arrest and increased replication fork stalling in AML samples ex vivo, and VE-822 potentiates the cytotoxicity of gemcitabine in an orthotopic mouse model of AML [101]. Moreover, ATRi in combination with splicing modulation show augmented efficacy in CD34+ cells with splicing factor mutations [73].
As the downstream effector of ATRi, CHK1 is serine/threonine kinase that promotes cycle arrest in response to DNA damage or replication stalling/stress [121]. A CHK1 inhibitor (CHK1i) UCN-01 can selectively induce apoptosis of cells with s SF3B1 mutation through cell cycle arrest, and increase the efficacy of the Sudemycin D6 when used together in the treatment of MDS/AML [14]. The nucleoside analog cytarabine is a potent and widely used drug in AML [122]. Etoposide inhibits topoisomerase II and induces cell cycle arrest, apoptosis, and autophagy [123]. Another small chemical SCH900776 was shown to disrupt cytarabine-induced CHK1 activation, leading to S phase arrest and significantly increased apoptosis in AML cell lines [104]. CHK1i also enhanced the effect of CPX-351 (liposomally encapsulated combination of cytarabine and daunorubicin) in AML cell lines and primary AML cells [124]. Moreover, the combination of ATRi and CHK1i shows synergistic suppression of ex vivo AML cell proliferation [103,125]. The elevated expression of cytosine deaminase APOBEC3A sensitizes AML cells to VE-822 and CHK1i of PF477736, thereby could be used as a biomarker for ATRi/CHK1i therapy of AML [102]. Additionally, combined inhibition of CHK1 and BCL-2 by LY2603618 and venetoclax synergistically induced apoptosis in AML cell lines and primary patient samples [105].
The tyrosine kinase Wee1 is a dual specificity kinase that phosphorylates CDK1 at Tyr15 to inhibit its kinase activity [126]. In response to DNA damage in the G2 phase, CHK1 phosphorylates the kinase WEE1 and the phosphatase CDC25C, resulting in CDK1 inhibition and cell cycle arrest at the G2/M checkpoint [121,126]. Combinatory inhibition of WEE1 and ATR/CHK1 showed a synergistic inhibitory effect in AML cells ex vivo [103,125]. In AML patient samples with SRSF2 mutation, WEE1 inhibitor adavosertib increases the vulnerability of mutant cells to CHK1i, implying the potential of combined therapy for MDS/AML with SRSF2 mutations [127].
4. Targeting Immune Responses and Inflammation Pathways
MDS is associated with immune dysfunction. On one hand, the activated innate immune system leads to abnormal hematopoiesis and unbalanced cell death and proliferation by directly affecting cytokine levels and activity of inflammatory pathways and immune cells. On the other hand, the adaptive immune system is activated by the expansion of malignant MDS stem cells, resulting in suppressed hematopoiesis and escaping from tumor surveillance. Due to this correlation, several immuno-suppressive and -modulatory therapies have been examined in selected MDS patients. Of note, LR-MDS has stronger inflammatory and cytotoxic features than HR-MDS, which is associated with a more suppressive microenvironment [82]. Moreover, the secretion profiles of cytokines are variable between different types of MDS; therefore, the treatment for each type of MDS, such as LR-MDS and HR-MDS, needs to be considered separately. For instance, the goal of treatment for LR-MDS is to correct peripheral cytopenia, whereas for patients with HR-MDS the primary target is the malignant clones [128].
A few small molecules targeting proinflammatory cytokines have been tested for the treatment of MDS patients, especially in LR-MDS. Luspatercept binds to TGF superfamily ligands and suppresses SMAD2/3 signaling. It was approved by FDA and EMA for transfusion-dependent LR-MDS patients with RS and/or SF3B1 mutation after treatment with erythropoiesis-stimulating agents, and showed higher response rates [102,103,107,108,125,129]. For HR-MDS, IRAK4-L promotes leukemogenesis through NF-κB signaling [17,38] and a small molecule CA-4948 of oral IRAK4 inhibitor is currently evaluated in a phase 1 trial for HR-MDS and AML (NCT04278768) [106]. Results from 14 patients with SF3B1, U2AF1, or FLT3 mutations were promising. Out of five AML patients, two reached complete remission/complete remission with partial hematologic recovery (CR/CRh) (1 CR, 1 CRh), and out of seven patients with spliceosome-mutated HR-MDS, four reached marrow CR [130]. The CA-4948 monotherapy was well tolerated and resulted in a reduction in bone marrow blasts in 89% of evaluated patients, and three patients with splicing factor mutations all achieved CR, and patients with objective responses also showed signs of hematologic recovery [131]. CA-4948 not only inhibits IRAK4 but also suppress FLT3-ITD AML progression in in vitro and in vivo models. It has shown safety and activity in patients with relapsed or refractory Non-Hodgkin Lymphomas and is now evaluated in patients with HR-MDS and AML [106].
In contrast, while many downstream splicing events have been reported to be altered due to splicing factor mutations in animal models and patient samples, including the important EZH2 gene [4,9], no gene other than IRAK4 has been shown to be a potential target for therapeutic purpose. Due to the complex immune responses in MDS/AML patients, further investigations should explore the immune response landscapes to search for specific targets. Clinical evaluations of individual treatments for LR-MDS and HR-MDS require careful consideration in future studies.
This entry is adapted from the peer-reviewed paper 10.3390/biomedicines10081972
This entry is offline, you can click here to edit this entry!