TAR-DNA binding protein-43 (TDP-43) is a ubiquitously expressed RNA binding protein with the capacity to bind over 6000 RNA and DNA targets—particularly those involved in RNA, mitochondrial, and lipid metabolism. The specific functions of TDP-43 include mRNA stabilisation, transcription, translation, splicing, axonal transport, apoptosis, microRNA processing, epigenetic modifications, and cryptic exon inclusion/repression. It is without doubt that any alteration to these structural domains and/or normal functions of TDP-43 could cause downstream destabilisation effects of the intended target(s). This entry discusses briefly the structure of the TDP-43 protein, and some of their functional impact in amyotrophic lateral sclerosis (ALS).
- TDP-43
- RNA
- mitochondria
1. Introduction
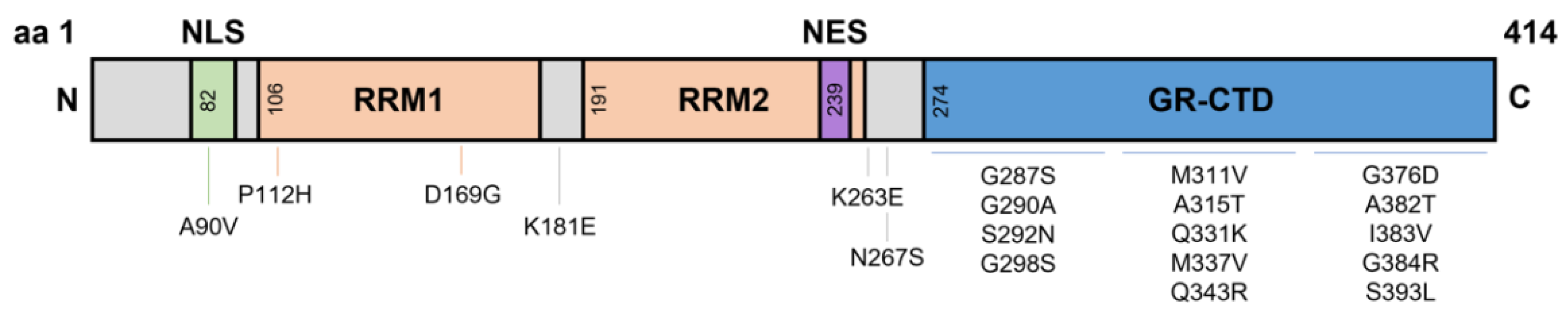
2. The N-Terminal Domain
3. RNA Recognition Motifs
4. Nuclear Localisation and Export Signals
5. Glycine-Rich C-Terminal Domain
This entry is adapted from the peer-reviewed paper 10.3390/metabo12080709
References
- Lagier-Tourenne, C.; Polymenidou, M.; Cleveland, D.W. TDP-43 and FUS/TLS: Emerging roles in RNA processing and neurodegeneration. Hum. Mol. Genet. 2010, 19, R46–R64.
- Buratti, E.; Baralle, F.E. TDP-43: Gumming up neurons through protein–protein and protein–RNA interactions. Trends Biochem. Sci. 2012, 37, 237–247.
- Wang, P.; Deng, J.; Dong, J.; Liu, J.; Bigio, E.H.; Mesulam, M.; Wang, T.; Sun, L.; Wang, L.; Lee, A.Y.-L. TDP-43 induces mitochondrial damage and activates the mitochondrial unfolded protein response. PLoS Genet. 2019, 15, e1007947.
- Lattante, S.; Rouleau, G.A.; Kabashi, E. TARDBP and FUS mutations associated with amyotrophic lateral sclerosis: Summary and update. Hum. Mutat. 2013, 34, 812–826.
- Kabashi, E.; Valdmanis, P.N.; Dion, P.; Spiegelman, D.; McConkey, B.J.; Velde, C.V.; Bouchard, J.-P.; Lacomblez, L.; Pochigaeva, K.; Salachas, F. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008, 40, 572–574.
- Daoud, H.; Valdmanis, P.N.; Kabashi, E.; Dion, P.; Dupre, N.; Camu, W.; Meininger, V.; Rouleau, G.A. Contribution of TARDBP mutations to sporadic amyotrophic lateral sclerosis. J. Med. Genet. 2009, 46, 112–114.
- Sreedharan, J.; Blair, I.P.; Tripathi, V.B.; Hu, X.; Vance, C.; Rogelj, B.; Ackerley, S.; Durnall, J.C.; Williams, K.L.; Buratti, E. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008, 319, 1668–1672.
- Wobst, H.J.; Delsing, L.; Brandon, N.J.; Moss, S.J. Truncation of the TAR DNA-binding protein 43 is not a prerequisite for cytoplasmic relocalization, and is suppressed by caspase inhibition and by introduction of the A90V sequence variant. PLoS ONE 2017, 12, e0177181.
- Chiò, A.; Mazzini, L.; D’Alfonso, S.; Corrado, L.; Canosa, A.; Moglia, C.; Manera, U.; Bersano, E.; Brunetti, M.; Barberis, M. The multistep hypothesis of ALS revisited: The role of genetic mutations. Neurology 2018, 91, e635–e642.
- Kuo, P.-H.; Chiang, C.-H.; Wang, Y.-T.; Doudeva, L.G.; Yuan, H.S. The crystal structure of TDP-43 RRM1-DNA complex reveals the specific recognition for UG-and TG-rich nucleic acids. Nucleic Acids Res. 2014, 42, 4712–4722.
- Kuo, P.-H.; Doudeva, L.G.; Wang, Y.-T.; Shen, C.-K.J.; Yuan, H.S. Structural insights into TDP-43 in nucleic-acid binding and domain interactions. Nucleic Acids Res. 2009, 37, 1799–1808.
- Buratti, E. TDP-43 post-translational modifications in health and disease. Expert Opin. Ther. Targets 2018, 22, 279–293.
- Costessi, L.; Porro, F.; Iaconcig, A.; Muro, A.F. TDP-43 regulates β-adducin (Add2) transcript stability. RNA Biol. 2014, 11, 1280–1290.
- Avendaño-Vázquez, S.E.; Dhir, A.; Bembich, S.; Buratti, E.; Proudfoot, N.; Baralle, F.E. Autoregulation of TDP-43 mRNA levels involves interplay between transcription, splicing, and alternative polyA site selection. Genes Dev. 2012, 26, 1679–1684.
- Buratti, E.; Baralle, F.E. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front. Biosci. 2008, 13, 867–878.
- Freibaum, B.D.; Chitta, R.K.; High, A.A.; Taylor, J.P. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J. Proteome Res. 2010, 9, 1104–1120.
- De Conti, L.; Akinyi, M.V.; Mendoza-Maldonado, R.; Romano, M.; Baralle, M.; Buratti, E. TDP-43 affects splicing profiles and isoform production of genes involved in the apoptotic and mitotic cellular pathways. Nucleic Acids Res. 2015, 43, 8990–9005.
- Alami, N.H.; Smith, R.B.; Carrasco, M.A.; Williams, L.A.; Winborn, C.S.; Han, S.S.; Kiskinis, E.; Winborn, B.; Freibaum, B.D.; Kanagaraj, A. Axonal transport of TDP-43 mRNA granules is impaired by ALS-causing mutations. Neuron 2014, 81, 536–543.
- Kawahara, Y.; Mieda-Sato, A. TDP-43 promotes microRNA biogenesis as a component of the Drosha and Dicer complexes. Proc. Natl. Acad. Sci. USA 2012, 109, 3347–3352.
- Pacetti, M.; De Conti, L.; Marasco, L.E.; Romano, M.; Rashid, M.M.; Nubiè, M.; Baralle, F.E.; Baralle, M. Physiological tissue-specific and age-related reduction of mouse TDP-43 levels is regulated by epigenetic modifications. Dis. Models Mech. 2022, 15, dmm049032.
- Ma, X.R.; Prudencio, M.; Koike, Y.; Vatsavayai, S.C.; Kim, G.; Harbinski, F.; Briner, A.; Rodriguez, C.M.; Guo, C.; Akiyama, T. TDP-43 represses cryptic exon inclusion in the FTD–ALS gene UNC13A. Nature 2022, 603, 124–130.
- Buratti, E.; Dörk, T.; Zuccato, E.; Pagani, F.; Romano, M.; Baralle, F.E. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. EMBO J. 2001, 20, 1774–1784.
- Shiina, Y.; Arima, K.; Tabunoki, H.; Satoh, J.-I. TDP-43 dimerizes in human cells in culture. Cell. Mol. Neurobiol. 2010, 30, 641–652.
- Zhang, Y.-J.; Caulfield, T.; Xu, Y.-F.; Gendron, T.F.; Hubbard, J.; Stetler, C.; Sasaguri, H.; Whitelaw, E.C.; Cai, S.; Lee, W.C. The dual functions of the extreme N-terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 2013, 22, 3112–3122.
- Afroz, T.; Hock, E.-M.; Ernst, P.; Foglieni, C.; Jambeau, M.; Gilhespy, L.A.; Laferriere, F.; Maniecka, Z.; Plückthun, A.; Mittl, P. Functional and dynamic polymerization of the ALS-linked protein TDP-43 antagonizes its pathologic aggregation. Nat. Commun. 2017, 8, 45.
- Jiang, L.-L.; Xue, W.; Hong, J.-Y.; Zhang, J.-T.; Li, M.-J.; Yu, S.-N.; He, J.-H.; Hu, H.-Y. The N-terminal dimerization is required for TDP-43 splicing activity. Sci. Rep. 2017, 7, 6196.
- Wang, A.; Conicella, A.E.; Schmidt, H.B.; Martin, E.W.; Rhoads, S.N.; Reeb, A.N.; Nourse, A.; Ramirez Montero, D.; Ryan, V.H.; Rohatgi, R. A single N-terminal phosphomimic disrupts TDP-43 polymerization, phase separation, and RNA splicing. EMBO J. 2018, 37, e97452.
- Vivoli-Vega, M.; Guri, P.; Chiti, F.; Bemporad, F. Insight into the folding and dimerization mechanisms of the N-terminal domain from human TDP-43. Int. J. Mol. Sci. 2020, 21, 6259.
- Tziortzouda, P.; Van Den Bosch, L.; Hirth, F. Triad of TDP43 control in neurodegeneration: Autoregulation, localization and aggregation. Nat. Rev. Neurosci. 2021, 22, 197–208.
- Mompeán, M.; Romano, V.; Pantoja-Uceda, D.; Stuani, C.; Baralle, F.E.; Buratti, E.; Laurents, D.V. Point mutations in the N-terminal domain of transactive response DNA-binding protein 43 kDa (TDP-43) compromise its stability, dimerization, and functions. J. Biol. Chem. 2017, 292, 11992–12006.
- Buratti, E.; Baralle, F.E. Characterization and functional implications of the RNA binding properties of nuclear factor TDP-43, a novel splicing regulator ofCFTR Exon 9. J. Biol. Chem. 2001, 276, 36337–36343.
- Tollervey, J.R.; Curk, T.; Rogelj, B.; Briese, M.; Cereda, M.; Kayikci, M.; König, J.; Hortobágyi, T.; Nishimura, A.L.; Župunski, V. Characterizing the RNA targets and position-dependent splicing regulation by TDP-43. Nat. Neurosci. 2011, 14, 452–458.
- Lukavsky, P.J.; Daujotyte, D.; Tollervey, J.R.; Ule, J.; Stuani, C.; Buratti, E.; Baralle, F.E.; Damberger, F.F.; Allain, F.H. Molecular basis of UG-rich RNA recognition by the human splicing factor TDP-43. Nat. Struct. Mol. Biol. 2013, 20, 1443–1449.
- Mackness, B.C.; Tran, M.T.; McClain, S.P.; Matthews, C.R.; Zitzewitz, J.A. Folding of the RNA recognition motif (RRM) domains of the amyotrophic lateral sclerosis (ALS)-linked protein TDP-43 reveals an intermediate state. J. Biol. Chem. 2014, 289, 8264–8276.
- Bose, J.K.; Wang, I.-F.; Hung, L.; Tarn, W.-Y.; Shen, C.-K.J. TDP-43 Overexpression Enhances Exon 7 Inclusion during the Survival of Motor Neuron Pre-mRNA Splicing. J. Biol. Chem. 2008, 283, 28852–28859.
- Chen, H.-J.; Topp, S.D.; Hui, H.S.; Zacco, E.; Katarya, M.; McLoughlin, C.; King, A.; Smith, B.N.; Troakes, C.; Pastore, A. RRM adjacent TARDBP mutations disrupt RNA binding and enhance TDP-43 proteinopathy. Brain 2019, 142, 3753–3770.
- Huang, Y.-C.; Lin, K.-F.; He, R.-Y.; Tu, P.-H.; Koubek, J.; Hsu, Y.-C.; Huang, J.J.-T. Inhibition of TDP-43 aggregation by nucleic acid binding. PLoS ONE 2013, 8, e64002.
- Liu, W.; Li, C.; Shan, J.; Wang, Y.; Chen, G. Insights into the aggregation mechanism of RNA recognition motif domains in TDP-43: A theoretical exploration. R. Soc. Open Sci. 2021, 8, 210160.
- Shodai, A.; Morimura, T.; Ido, A.; Uchida, T.; Ayaki, T.; Takahashi, R.; Kitazawa, S.; Suzuki, S.; Shirouzu, M.; Kigawa, T. Aberrant assembly of RNA recognition motif 1 links to pathogenic conversion of TAR DNA-binding protein of 43 kDa (TDP-43). J. Biol. Chem. 2013, 288, 14886–14905.
- Chang, C.-K.; Chiang, M.-H.; Toh, E.K.-W.; Chang, C.-F.; Huang, T.-H. Molecular mechanism of oxidation-induced TDP-43 RRM1 aggregation and loss of function. FEBS Lett. 2013, 587, 575–582.
- Colombrita, C.; Zennaro, E.; Fallini, C.; Weber, M.; Sommacal, A.; Buratti, E.; Silani, V.; Ratti, A. TDP-43 is recruited to stress granules in conditions of oxidative insult. J. Neurochem. 2009, 111, 1051–1061.
- Ayala, Y.M.; Zago, P.; D’Ambrogio, A.; Xu, Y.-F.; Petrucelli, L.; Buratti, E.; Baralle, F.E. Structural determinants of the cellular localization and shuttling of TDP-43. J. Cell Sci. 2008, 121, 3778–3785.
- Yang, C.; Qiao, T.; Yu, J.; Wang, H.; Guo, Y.; Salameh, J.; Metterville, J.; Parsi, S.; Yusuf, I.; Brown, R.H. Low-level overexpression of wild type TDP-43 causes late-onset, progressive neurodegeneration and paralysis in mice. PLoS ONE 2022, 17, e0255710.
- Winton, M.J.; Igaz, L.M.; Wong, M.M.; Kwong, L.K.; Trojanowski, J.Q.; Lee, V.M.-Y. Disturbance of nuclear and cytoplasmic TAR DNA-binding protein (TDP-43) induces disease-like redistribution, sequestration, and aggregate formation. J. Biol. Chem. 2008, 283, 13302–13309.
- Stade, K.; Ford, C.S.; Guthrie, C.; Weis, K. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 1997, 90, 1041–1050.
- Pinarbasi, E.S.; Cağatay, T.; Fung, H.Y.J.; Li, Y.C.; Chook, Y.M.; Thomas, P.J. Active nuclear import and passive nuclear export are the primary determinants of TDP-43 localization. Sci. Rep. 2018, 8, 7083.
- Buratti, E.; Brindisi, A.; Giombi, M.; Tisminetzky, S.; Ayala, Y.M.; Baralle, F.E. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: An important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. J. Biol. Chem. 2005, 280, 37572–37584.
- Suk, T.R.; Rousseaux, M.W. The role of TDP-43 mislocalization in amyotrophic lateral sclerosis. Mol. Neurodegener. 2020, 15, 45.
- Ayala, Y.M.; De Conti, L.; Avendaño-Vázquez, S.E.; Dhir, A.; Romano, M.; D’ambrogio, A.; Tollervey, J.; Ule, J.; Baralle, M.; Buratti, E. TDP-43 regulates its mRNA levels through a negative feedback loop. EMBO J. 2011, 30, 277–288.
- Mercado, P.A.; Ayala, Y.M.; Romano, M.; Buratti, E.; Baralle, F.E. Depletion of TDP 43 overrides the need for exonic and intronic splicing enhancers in the human apoA-II gene. Nucleic Acids Res. 2005, 33, 6000–6010.
- Udan, M.; Baloh, R.H. Implications of the prion-related Q/N domains in TDP-43 and FUS. Prion 2011, 5, 1–5.
- Sun, Y.; Chakrabartty, A. Phase to phase with TDP-43. Biochemistry 2017, 56, 809–823.
- Monahan, Z.; Shewmaker, F.; Pandey, U.B. Stress granules at the intersection of autophagy and ALS. Brain Res. 2016, 1649, 189–200.
- Nishimoto, Y.; Ito, D.; Yagi, T.; Nihei, Y.; Tsunoda, Y.; Suzuki, N. Characterization of alternative isoforms and inclusion body of the TAR DNA-binding protein-43. J. Biol. Chem. 2010, 285, 608–619.
- Hong, K.; Li, Y.; Duan, W.; Guo, Y.; Jiang, H.; Li, W.; Li, C. Full-length TDP-43 and its C-terminal fragments activate mitophagy in NSC34 cell line. Neurosci. Lett. 2012, 530, 144–149.
- Liu-Yesucevitz, L.; Bilgutay, A.; Zhang, Y.-J.; Vanderwyde, T.; Citro, A.; Mehta, T.; Zaarur, N.; McKee, A.; Bowser, R.; Sherman, M. Tar DNA binding protein-43 (TDP-43) associates with stress granules: Analysis of cultured cells and pathological brain tissue. PLoS ONE 2010, 5, e13250.
- Igaz, L.M.; Kwong, L.K.; Xu, Y.; Truax, A.C.; Uryu, K.; Neumann, M.; Clark, C.M.; Elman, L.B.; Miller, B.L.; Grossman, M. Enrichment of C-terminal fragments in TAR DNA-binding protein-43 cytoplasmic inclusions in brain but not in spinal cord of frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Am. J. Pathol. 2008, 173, 182–194.
- Zhang, Y.-J.; Xu, Y.-F.; Cook, C.; Gendron, T.F.; Roettges, P.; Link, C.D.; Lin, W.-L.; Tong, J.; Castanedes-Casey, M.; Ash, P. Aberrant cleavage of TDP-43 enhances aggregation and cellular toxicity. Proc. Natl. Acad. Sci. USA 2009, 106, 7607–7612.
- Che, M.-X.; Jiang, L.-L.; Li, H.-Y.; Jiang, Y.-J.; Hu, H.-Y. TDP-35 sequesters TDP-43 into cytoplasmic inclusions through binding with RNA. FEBS Lett. 2015, 589, 1920–1928.
- Che, M.X.; Jiang, Y.J.; Xie, Y.Y.; Jiang, L.L.; Hu, H.Y. Aggregation of the 35-kDa fragment of TDP-43 causes formation of cytoplasmic inclusions and alteration of RNA processing. FASEB J. 2011, 25, 2344–2353.
- Xiao, S.; Sanelli, T.; Chiang, H.; Sun, Y.; Chakrabartty, A.; Keith, J.; Rogaeva, E.; Zinman, L.; Robertson, J. Low molecular weight species of TDP-43 generated by abnormal splicing form inclusions in amyotrophic lateral sclerosis and result in motor neuron death. Acta Neuropathol. 2015, 130, 49–61.
- Tsuji, H.; Arai, T.; Kametani, F.; Nonaka, T.; Yamashita, M.; Suzukake, M.; Hosokawa, M.; Yoshida, M.; Hatsuta, H.; Takao, M. Molecular analysis and biochemical classification of TDP-43 proteinopathy. Brain 2012, 135, 3380–3391.