1. Introduction
TDP-43 is an essential protein involved in RNA processing [
7], cellular metabolism [
8], and mitochondrial function [
9]. It is widely accepted that metabolic processes (both cellular and systemic) are dysregulated in ALS patients and disease relevant models. The majority of
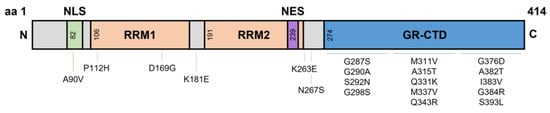
TARDBP mutations reside in the hyper-mutable glycine-rich C-terminal domain, with other mutations spread across the different domains such as the RNA recognition motifs (P112H, D169G, K181E) and the nuclear localisation signal (A90V)—with potentially several more mutations yet to be identified [
10,
11,
12] (
Figure 1). Several groups have generated models with rare
TARDBP mutations to understand their contribution towards protein misfolding and neuronal toxicity in ALS. In vitro studies of the C-terminal Q331K and G294A mutations identified a higher propensity to form C-terminal fragments, leading to cell death; most often these mutations produced new phosphorylation sites or disrupted RNA binding sites [
13]. By contrast, the NLS domain A90V mutation results in cytoplasmic inclusions, however does not cause cell death in vitro [
14]. These studies suggest that although TDP-43 mutations can cause cytoplasmic aggregates, additional factors may be required for neuronal toxicity and subsequent motor neuron death; in line with the multi-step hypothesis of ALS [
15]. Given the large number of TDP-43 RNA targets, a disruption of normal TDP-43 could lead to disease-causing mechanisms such as RNA instability, oxidative stress, mitochondrial dysfunction, and even dysregulated lipid metabolism.
The
TAR-DNA binding protein (
TARDBP) gene encodes a highly conserved and ubiquitously expressed 43 kDa heterogenous nuclear ribonucleoprotein (hnRNP), TDP-43.
TARDBP is located on chromosome 1 and comprises of 6 exons forming 414 amino acids (aa). These six exons form domains that are bookended by an N-terminal domain (NTD) (aa1–76) and C-terminal domain (CTD) (aa274–414). Like many other hnRNPs, the domains within TDP-43 include a nuclear localisation signal (NLS aa82–98), two RNA recognition motifs (RRM1 aa106–176; RRM2 aa191–259), and a nuclear export signal (NES aa239–250) residing within RRM2 (
Figure 1) [
16,
17]. Additionally, specific sites are located within these domains for other functions such as caspase-3 cleavage sites, post-translational modification, and mitochondrial localisation (reviewed in Buratti [
18]). Ultimately, the unique structure of TDP-43 gives rise to its capacity to bind and regulate over 6000 RNA species, in addition to its own autoregulation abilities. The specific functions of TDP-43 include mRNA stabilisation [
19], transcription [
20,
21], translation [
22], splicing [
21,
22,
23], axonal transport [
24], apoptosis [
23], microRNA processing [
25], epigenetic modifications [
26] and cryptic exon inclusion/repression [
27,
28]. It is without doubt that any alteration to these structural domains and/or normal functions of TDP-43 could cause downstream destabilisation effects of the intended target(s).
2. The N-Terminal Domain
The main functions of the NTD are to self-regulate
TARDBP mRNA through homodimerisation and aid mRNA splicing [
29,
30,
31,
32,
33]. TDP-43 can form homodimers, spanning a spectrum of oligomeric species with dynamic folding states [
31,
34]. NTD generated TDP-43 oligomers do not interact with other RNA like proteins such as hnRNPA1/2 and are distinct from stress or inclusion formed cytoplasmic oligomers [
30,
31]. It is speculated that NTD generated oligomers adopt structures resistant to cellular stress and act as preventative measures against cytoplasmic inclusion formation under normal conditions [
32,
35]. However, in a stressed HEK293T TDP-43 overexpression model, these NTD oligomers were aggregated within large cytoplasmic inclusions, apparently yielding to cellular stress [
29]. Despite this, under physiological conditions there is debate surrounding the importance and function of these oligomers. Avendaño-Vázquez et al., has suggested that a dimer equilibrium is required for proper TDP-43 function [
20]. TDP-43 protein missing the NTD displays significant β-sheet folding abnormalities, consequently leading to a loss of TDP-43 function [
30,
32]. Interestingly plasmids missing just the first 10 amino acid residues have been shown to exhibit similar significant β-sheet folding abnormalities and structural destabilisation, suggesting the first 10 amino acids are the most crucial of the 76 in the NTD [
30,
32]. Additionally, the NTD of TDP-43 is known to enhance exon 9 inclusion of the
CFTR gene; should exon 9 be excluded, patients develop cystic fibrosis [
28]. In several in vitro models inducing NTD mutations, TDP-43 was unable to repress aberrant splicing of exon 9 in
CFTR, indicating the importance of the NTD in regulating splicing [
30,
32,
33]. In another in vitro study, point mutations at these NTD sites: V31R, T32R, and L28A resulted in similar incomplete TDP-43 folding, abolished splicing activity, as well as induced cytoplasmic inclusions [
36]. Taken together, these studies demonstrate the importance of NTD oligomers for proper folding, splicing, RNA metabolism, and in preventing the formation of cytoplasmic aggregates [
31,
34].
3. RNA Recognition Motifs
RRMs direct RNA and DNA binding. TDP-43 RRMs sit in tandem and can bind to over 6000 targets, with a 20–60-fold higher binding affinity for long UG repeats [
37,
38,
39,
40]. These RRMs are separated by a unique 15 amino acid cross-linker spanning four β-strands, allowing for additional interactions between TDP-43 and other RNAs [
39]. The RRM domains of TDP-43 enable splicing control [
41,
42], pre-mRNA regulation [
43], physiological oligomer formation [
44,
45], and possibly aggregate clearing [
43,
44,
46]. Although these RRMs are similar in sequence, they form different tertiary structures, where RRM1 preferentially binds longer (UG
6) repeats, and RRM2 binds shorter (UG
3) repeats [
39]. Interestingly, these RRMs are prone to oxidative stress due to the occurrence of cysteine residues; these residues are accessible on the β-loop of RRM1, however are buried and inaccessible on RRM2 [
46]. Due to these structural differences, the RRM2 domain are able to form highly stable assemblies with DNA strands, although it is unknown whether these assemblies contribute to cytoplasmic aggregations [
17]. It has been demonstrated that these RRMs induce oligomer formation as a means to prevent TDP-43 aggregation [
44]. However, when cysteine residues on RRM1 are oxidised they undergo oligomer conformational changes resulting in TDP-43 insoluble aggregates [
46]. In an NSC-34 stressed model, oligomer formation was disrupted, leading to binding with RNAs destined for stress granule formation [
47]. In a HEK293T TDP-43
K181E mutation model the structure of TDP-43 and its RRMs were unaffected; however phosphorylated and ubiquitinated TDP-43 increased 4-fold [
42]. Upon further examination, these phosphorylated TDP-43 also sequestered healthy TDP-43 into their inclusions suggesting a detrimental effect of mutated TDP-43 RRMs [
42]. Taken together, there is a scarcity of studies examining the role of RRMs and their effect on RNA metabolism and TDP-43 aggregation. Thus, further research is required to fully elucidate the role of RRMs in RNA metabolism, cellular toxicity, and its propensity to negatively react to oxidative stress, a key mechanism put forth for ALS.
4. Nuclear Localisation and Export Signals
Shuttling of TDP-43 to and from the nucleus and the cytoplasm is controlled by the NLS and NES domains [
48]. Under physiological conditions TDP-43 is predominantly localised to the nucleus and is exported to the cytoplasm for crucial activities such as mRNA stability and binding [
48]. In mouse in vitro (3T3) and in vivo models, overexpression of TDP-43
WT does not induce cytoplasmic inclusions, however TDP-43 was observed more frequently in the cytoplasm compared to the nucleus [
48,
49]. Nuclear localisation targeting is dependent on two clusters of basic residues around aa82–98 [
50]. When both clusters are mutated, impaired nuclear localisation occurs and extensive accumulation of TDP-43 cytoplasmic C-terminal fragments are observed [
32,
48,
50]. However, when only the first cluster is altered, TDP-43 is still able to localise to the nucleus [
48]. Evidently, redundancies are in place for nuclear localisation should one cluster become impaired.
It could also be possible that defective export may cause TDP-43 to remain in the nucleus, prohibiting normal mRNA capabilities within the cytoplasm. Most hnRNPs are exported by a broad cargo exportin (XPO1) [
51]. Previously, TDP-43 nuclear magnetic resonance and predictive modelling has been used to visualise binding domains of NES within the RRM [
52]. Interestingly, all potential binding sites of the NES are buried within the core of the RRMs, indicating that the NES is inaccessible for XPO1 binding [
52]. In line with this finding, when an NES plasmid was exposed to XPO1, only weak bonds formed; demonstrating that the NES may not be the main domain required for shuttling TDP-43 into the cytoplasm [
52]. Upon removal of the NES, similar amounts of TDP-43 were observed in the cytoplasm compared to wild-type, suggesting a ‘passive’ movement of TDP-43 to the cytoplasm in the absence of XPO1 binding [
52]. However, when inhibiting the NES, TDP-43 forms insoluble inclusions, suggesting that the NES is still required for RRM functions and proper export [
48,
52]. Despite limited research into the specific structure of TDP-43 NLS and NES domains, it is plausible that shuttling of TDP-43 is also dictated by the self-regulation or splicing of its RRMs and CTD. Other possibilities include other exportins such as exon junction complexes, nuclear export receptors, and THO/transcription export complexes which have not yet been investigated in TDP-43 models. Further investigations are still warranted to fully characterise the NES and NLS domains to understand how they might contribute to the accumulation of disease relevant TDP-43 pathology.
5. Glycine-Rich C-Terminal Domain
The main function of the glycine rich CTD is to mediate protein-protein interactions via hnRNP interactions [
53]. Other known functions include export and shuttling [
48,
54], gene transcription [
55], and splicing regulation [
56]. One way in which the glycine rich CTD modifies gene regulation is through the Q/N (asparagine) rich region, also known as the prion-like region [
57]. This prion-like region drives gene regulation through RNP granule assembly [
58]. These granules play a vital role in the stress granule response, however, should the clearance and reversibility of these granules fail, a high propensity for aggregation occurs [
59]. It has been suggested that disruptions to the nucleic-acid-binding capability and the removal of the C-terminal tail causes TDP-43 mislocalisation due to the improper return to the nucleus [
48].
The structure, function, and disruption of the CTD has mostly been studied in the context of ALS, due to its propensity to mutate. Most studies focus either on generating C-terminal fragments or introducing point mutations. Cleavage of TDP-43 via caspase3/7 at Asp89 and Asp174 generates 25 kDa or 35 kDa C-terminal fragment and a quickly degraded N-terminal fragment [
60,
61]. Due to RRM domain retention, these generated C-terminal fragments can bind to one another forming oligomers [
39]. A wide scope of evidence in in vitro and in vivo models indicate that neuronal toxicity is caused by C-terminal cytoplasmic inclusions [
62,
63], 25 kDa fragment overexpression [
64], and/or 35 kDa fragment overexpression [
65,
66,
67]. However, the demonstration of neuronal toxicity by these C-terminal fragments are scarce and remain poorly understood. What we do know is that the 35 kDa fragment alters pre-mRNA splicing, sequesters full length TDP-43 at RRM1, and forms oligomer stress granules via TDP-43 recruitment [
65,
66,
67]. On the other hand, it has been shown that the 25 kDa fragment in a HEK293T model induces cell death, despite being unable to alter or bind to full length TDP-43 [
64]. It is clear that neuronal toxicity occurs; however, the mechanism is unknown and more importantly remains elusive for individuals lacking
TARDBP mutations given that the 25 kDa fragment is most commonly identified in post-mortem studies of ALS patients [
67,
68]. It is likely that a greater understanding of other post-translational modifications involving phosphorylation, caspases, or even oxidative stress may aide in our knowledge of cytoplasmic TDP-43 induced neuronal toxicity in ALS.
This entry is adapted from the peer-reviewed paper 10.3390/metabo12080709