Eryptosis is a coordinated, programmed cell death culminating with the disposal of cells without disruption of the cell membrane and the release of endocellular oxidative and pro-inflammatory milieu. While providing a convenient form of death for erythrocytes, dysregulated eryptosis may result in a series of detrimental and harmful pathological consequences highly related to the endothelial dysfunction (ED). Metabolic syndrome (MetS) is described as a cluster of cardiometabolic factors (hyperglycemia, dyslipidemia, hypertension and obesity) that increases the risk of cardiovascular complications such as those related to diabetes and atherosclerosis.
- eryptosis
- metabolic syndrome
- diabetes
- dyslipidemia
- hypertension
- obesity
- atherosclerosis
- vascular damage
- oxidative stress
- endothelial dysfunction
1. Eryptosis
1.1. Mechanisms
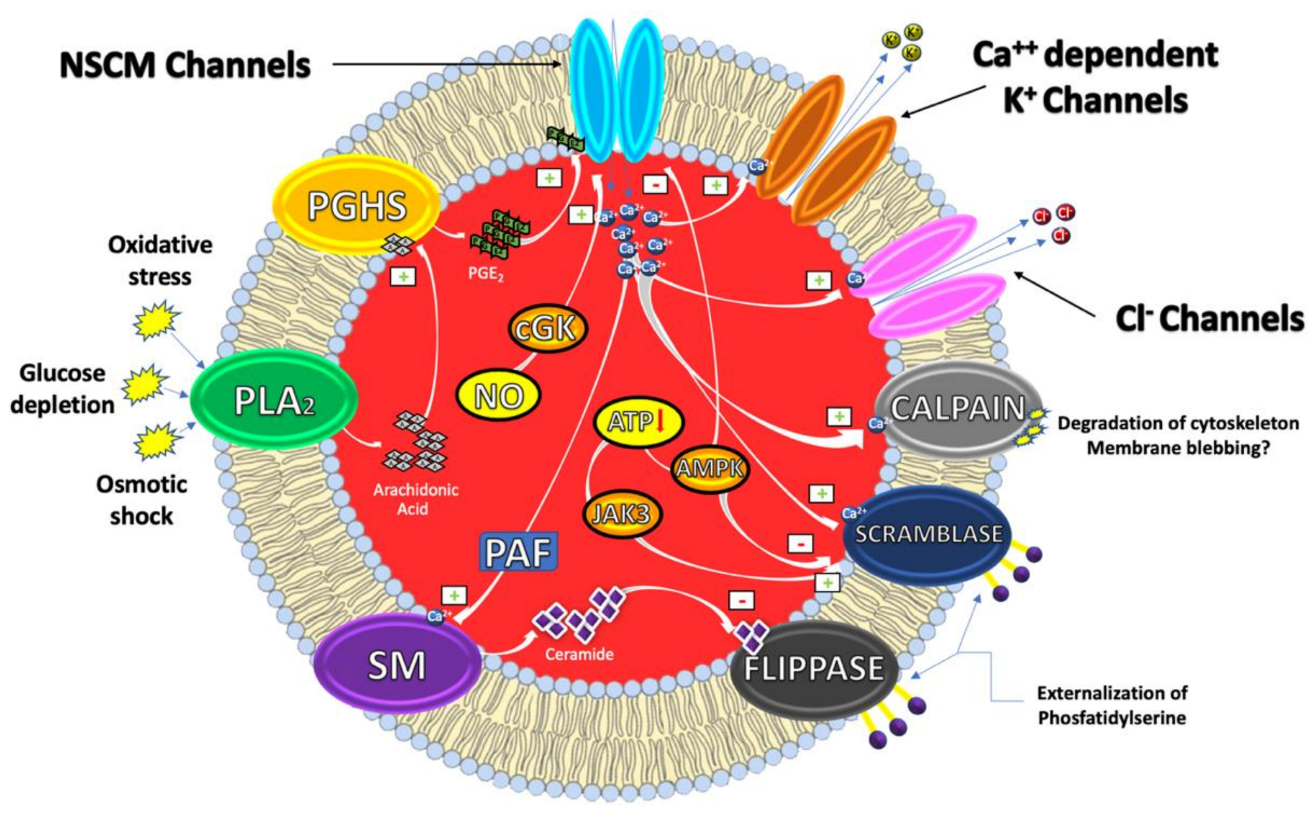
2. Eryptosis, Hyperglycemia and Diabetes
3. Eryptosis, Dyslipidemia and Atherosclerosis
This entry is adapted from the peer-reviewed paper 10.3390/antiox10020154
References
- Bosman, G.J.C.G.M.; Willekens, F.L.A.; Werre, J.M. Erythrocyte aging: A more than superficial resemblance to apoptosis? Cell. Physiol. Biochem. 2005, 16, 1–8.
- Arese, P.; Turrini, F.; Schwarzer, E. Band 3/complement-mediated recognition and removal of normally senescent and pathological human erythrocytes. Cell. Physiol. Biochem. 2005, 16, 133–146.
- Lang, F.; Qadri, S.M. Mechanisms and significance of eryptosis, the suicidal death of erythrocytes. Blood Purif. 2012, 33, 125–130.
- Lang, E.; Qadri, S.M.; Lang, F. Killing me softly—Suicidal erythrocyte death. Int. J. Biochem. Cell Biol. 2012, 44, 1236–1243.
- Haus, J.M.; Kashyap, S.R.; Kasumov, T.; Zhang, R.; Kelly, K.R.; Defronzo, R.A.; Kirwan, J.P. Plasma ceramides are elevated in obese subjects with type 2 diabetes and correlate with the severity of insulin resistance. Diabetes 2009, 58.
- Lang, F.; Gulbins, E.; Lang, P.A.; Zappulla, D.; Föller, M. Ceramide in suicidal death of erythrocytes. Cell. Physiol. Biochem. 2010, 26, 21–28.
- Adams, J.M.; Pratipanawatr, T.; Berria, R.; Wang, E.; DeFronzo, R.A.; Sullards, M.C.; Mandarino, L.J. Ceramide Content Is Increased in Skeletal Muscle from Obese Insulin-Resistant Humans. Diabetes 2004, 53.
- Lang, E.; Lang, F. Triggers, inhibitors, mechanisms, and significance of eryptosis: The suicidal erythrocyte death. Biomed. Res. Int. 2015.
- Repsold, L.; Joubert, A.M. Eryptosis: An Erythrocyte’s Suicidal Type of Cell Death. Biomed. Res. Int. 2018.
- Pretorius, E.; Du Plooy, J.N.; Bester, J. A Comprehensive Review on Eryptosis. Cell. Physiol. Biochem. 2016, 39, 1977–2000.
- Lang, F.; Abed, M.; Lang, E.; Föller, M. Oxidative stress and suicidal erythrocyte death. Antioxid. Redox Signal. 2014, 21.
- Qadri, S.M.; Bissinger, R.; Solh, Z.; Oldenborg, P.A. Eryptosis in health and disease: A paradigm shift towards understanding the (patho) physiological implications of programmed cell death of erythrocytes. Blood Rev. 2017, 31, 349–361.
- Lang, E.; Lang, F. Mechanisms and pathophysiological significance of eryptosis, the suicidal erythrocyte death. Semin. Cell Dev. Biol. 2015, 39, 35–42.
- Brownlee, M. Biochemistry and molecular cell biology of diabetic complications. Nature 2001, 414, 813–820.
- Giacco, F.; Brownlee, M. Oxidative Stress and Diabetic Complications. Circ. Res. 2010, 107, 1058–1070.
- Webster, A.C.; Nagler, E.V.; Morton, R.L.; Masson, P. Chronic Kidney Disease. Lancet 2017, 389, 1238–1252.
- Lam, C.S.Y.; Benzie, I.F.F.; Choi, S.W.; Chan, L.Y.L.; Yeung, V.T.F.; Woo, G.C. Relationships among diabetic retinopathy, antioxidants, and glycemic control. Optom. Vis. Sci. 2011, 88.
- Gorban de Lapertosa, S.; Fereira de Moura, A.; Decroux, C.; Duke, L.; Hammond, L.; Jacobs, E.; Kaundal, A.; Li, J.; Liu, J.; Ohlrogge, A.E.; et al. Diabetes Atlas, 9th ed.; International Diabetes Federation: Brussels, Belgium, 2019.
- Deray, G.; Heurtier, A.; Grimaldi, A.; Launay Vacher, V.; Isnard Bagnis, C. Anemia and diabetes. Am. J. Nephrol. 2004, 24, 522–526.
- Gauci, R.; Hunter, M.; Bruce, D.G.; Davis, W.A.; Davis, T.M.E. Anemia complicating type 2 diabetes: Prevalence, risk factors and prognosis. J. Diabetes Complic. 2017, 31.
- Singh, D.K.; Winocour, P.; Farrington, K. Erythropoietic stress and anemia in diabetes mellitus. Nat. Rev. Endocrinol. 2009, 5, 204–210.
- Calderón-Salinas, J.V.; Muñoz-Reyes, E.G.; Guerrero-Romero, J.F.; Rodríguez-Morán, M.; Bracho-Riquelme, R.L.; Carrera-Gracia, M.A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in type 2 diabetic mellitus patients with chronic kidney disease. Mol. Cell. Biochem. 2011, 357.
- Maellaro, E.; Leoncini, S.; Moretti, D.; Del Bello, B.; Tanganelli, I.; De Felice, C.; Ciccoli, L. Erythrocyte caspase-3 activation and oxidative imbalance in erythrocytes and in plasma of type 2 diabetic patients. Acta Diabetol. 2013, 50.
- Vlassopoulos, A.; Lean, M.E.J.; Combet, E. Role of oxidative stress in physiological albumin glycation: A neglected interaction. Free Radic. Biol. Med. 2013, 60.
- Huebschmann, A.G.; Regensteiner, J.G.; Vlassara, H.; Reusch, J.E.B. Diabetes and advanced glycoxidation end products. Diabetes Care 2006, 29, 1420–1432.
- Vlassara, H.; Striker, G.E. Advanced Glycation Endproducts in Diabetes and Diabetic Complications. Endocrinol. Metab. Clin. North. Am. 2013, 42, 697–719.
- Awasthi, S.; Gayathiri, S.K.; Ramya, R.; Duraichelvan, R.; Dhason, A.; Saraswathi, N.T. Advanced Glycation-Modified Human Serum Albumin Evokes Alterations in Membrane and Eryptosis in Erythrocytes. Appl. Biochem. Biotechnol. 2015, 177.
- Ahmed, N. Advanced glycation endproducts—Role in pathology of diabetic complications. Diabetes Res. Clin. Pract. 2005, 67, 3–21.
- Vlassara, H.; Uribarri, J. Advanced glycation end products (AGE) and diabetes: Cause, effect, or both? Curr. Diabetes Rep. 2014, 14.
- Nowotny, K.; Jung, T.; Höhn, A.; Weber, D.; Grune, T. Advanced glycation end products and oxidative stress in type 2 diabetes mellitus. Biomolecules 2015, 5, 194–222.
- Singh, R.; Barden, A.; Mori, T.; Beilin, L. Advanced glycation end-products: A review. Diabetologia 2001, 44, 129–146.
- Ikeda, K.; Higashi, T.; Sano, H.; Jinnouchi, Y.; Yoshida, M.; Araki, T.; Ueda, S.; Horiuchi, S. Ne-(carboxymethyl)lysine protein adduct is a major immunological epitope in proteins modified with advanced glycation end products of the maillard reaction. Biochemistry 1996, 35.
- Reddy, S.; Bichler, J.; Wells-Knecht, K.J.; Thorpe, S.R.; Baynes, J.W. Nε-(Carboxymethyl) lysine Is a Dominant Advanced Glycation End Product (AGE) Antigen in Tissue Proteins. Biochemistry 1995, 34.
- Rabbani, N.; Thornalley, P.J. The critical role of methylglyoxal and glyoxalase 1 in diabetic nephropathy. Diabetes 2014, 63, 50–52.
- Riboulet-Chavey, A.; Pierron, A.; Durand, I.; Murdaca, J.; Giudicelli, J.; Van Obberghen, E. Methylglyoxal impairs the insulin signaling pathways independently of the formation of intracellular reactive oxygen species. Diabetes 2006, 55.
- Matafome, P.; Sena, C.; Seiça, R. Methylglyoxal, obesity, and diabetes. Endocrine 2013, 43, 472–484.
- Beisswenger, P.J.; Howell, S.K.; Touchette, A.D.; Lal, S.; Szwergold, B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes 1999, 48.
- Kilhovd, B.K.; Giardino, I.; Torjesen, P.A.; Birkeland, K.I.; Berg, T.J.; Thornalley, P.J.; Brownlee, M.; Hanssen, K.F. Increased serum levels of the specific AGE-compound methylglyoxal-derived hydroimidazolone in patients with type 2 diabetes. Metabolism 2003, 33.
- Lapolla, A.; Flamini, R.; Dalla Vedova, A.; Senesi, A.; Reitano, R.; Fedele, D.; Basso, E.; Seraglia, R.; Traldi, P. Glyoxal and methylglyoxal levels in diabetic patients: Quantitative determination by a new GC/MS method. Clin. Chem. Lab. Med. 2003, 41.
- Nagai, R.; Deemer, E.K.; Brock, J.W.; Thorpe, S.R.; Baynes, J.W. Effect of glucose concentration on formation of AGEs in erythrocytes In Vitro. Ann. N. Y. Acad. Sci. 2005, 1043, 146–150.
- Nicolay, J.; Schneider, J.; Niemoeller, O.; Artunc, F.; Portero-Otin, M.; Haik, G.; Thornalley, P.; Schleicher, E.; Wieder, T.; Lang, F. Stimulation of suicidal erythrocyte death by methylglyoxal. Cell. Physiol. Biochem. 2006, 40.
- Thornalley, P.J.; Jahan, I.; Ng, R. Suppression of the accumulation of triosephosphates and increased formation of methylglyoxal in human red blood cells during hyperglycaemia by thiamine In Vitro. J. Biochem. 2001, 129.
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose In Vitro. Biochem. J. 1988, 254.
- Kempe-Teufel, D.S.; Bissinger, R.; Qadri, S.M.; Wagner, R.; Peter, A.; Lang, F. Cellular markers of eryptosis are altered in type 2 diabetes. Clin. Chem. Lab. Med. 2018, 56, e177–e180.
- Wali, R.K.; Jaffe, S.; Kumar, D.; Kalra, V.K. Alterations in organization of phospholipids in erythrocytes as factor in adherence to endothelial cells in diabetes mellitus. Diabetes 1988, 62.
- Vekic, J.; Zeljkovic, A.; Stefanovic, A.; Jelic-Ivanovic, Z.; Spasojevic-Kalimanovska, V. Obesity and dyslipidemia. Metabolism 2019, 92, 71–81.
- Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218–1240.
- Kopin, L.; Lowenstein, C. In the Clinic® dyslipidemia. Ann. Intern. Med. 2017, 167.
- Schnabel, R.B.; Baumert, J.; Barbalic, M.; Dupuis, J.; Ellinor, P.T.; Durda, P.; Dehghan, A.; Bis, J.C.; Illig, T.; Morrison, A.C.; et al. Duffy antigen receptor for chemokines (Darc) polymorphism regulates circulating concentrations of monocyte chemoattractant protein-1 and other inflammatory mediators. Blood 2010, 115.
- Kolodgie, F.D.; Gold, H.K.; Burke, A.P.; Fowler, D.R.; Kruth, H.S.; Weber, D.K.; Farb, A.; Guerrero, L.J.; Hayase, M.; Kutys, R.; et al. Intraplaque Hemorrhage and Progression of Coronary Atheroma. N. Engl. J. Med. 2003, 32.
- Virmani, R.; Roberts, W.C. Extravasated erythrocytes, iron, and fibrin in atherosclerotic plaques of coronary arteries in fatal coronary heart disease and their relation to luminal thrombus: Frequency and significance in 57 necropsy patients and in 2958 five mm segments of 224 majo. Am. Heart J. 1983, 105.
- Pinzón-Díaz, C.E.; Calderón-Salinas, J.V.; Rosas-Flores, M.M.; Hernández, G.; López-Betancourt, A.; Quintanar-Escorza, M.A. Eryptosis and oxidative damage in hypertensive and dyslipidemic patients. Mol. Cell. Biochem. 2018, 11.