Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Chronic myeloid leukaemia (CML) is a type of blood cancer that is currently well-managed with drugs that target cancer-causing proteins. However, a significant proportion of CML patients do not respond to those drug treatments or relapse when they stop those drugs because the cancer cells in those patients stop relying on that protein and instead develop a new way to survive. Therefore, new treatment strategies may be necessary for those patients.
- chronic myeloid leukaemia
- tyrosine kinase inhibitors
- therapy resistance
- BCR::ABL1-independent mechanism of TKI resistance
- combination treatments
1. Introduction
Chronic myeloid leukaemia (CML) is a malignancy characterized by the clonal proliferation of white blood cells that are mostly originated from the myeloid lineage in the bone marrow [1]. CML arises from the t(9;22)(q34;q11) balanced reciprocal translocation between chromosome 9 and 22 that forms the Philadelphia chromosome [2][3]. The translocation event results in the fusion of the Breakpoint Cluster Region (BCR) gene with the Abelson proto-oncogene 1 (ABL1) gene, generating the BCR::ABL1 fusion gene [4]. The resulting chimeric protein, BCR::ABL1, is a potent tyrosine-kinase signalling protein that drives cell proliferation and reduces apoptosis, which causes leukaemia [4]. Depending on the position of the BCR breakpoint, different BCR::ABL1 protein isoforms are generated (Figure 1A) [5]. The e13a2/e14a2 alternative transcripts (or b2a2/b3a2), resulting from the juxtaposition of BCR exon 13 or 14 with ABL1 exon 2, produce a 210 kDa protein, which is found in over 90% of CML patients. The e1a2 transcript encodes a 190 kDa protein (p190), which is rare in CML but is relatively common in acute lymphoblastic leukaemia, occurring in around 70% of cases (Figure 1B) [5][6]. Approximately 95% of CML patients are diagnosed at the chronic phase, which is relatively indolent but can involve symptoms such as fatigue, abdominal pain, or weight loss. During this phase, the disease can be effectively managed with tyrosine kinase inhibitors (TKIs). If untreated, CML could progress to an accelerated phase (AP), which can last for up to a year, and eventually progress into the terminal blast phase of the disease, termed the blast crisis (BC). The blast crisis is characterized by the presence of excess blast cells in the blood or bone marrow [7]. Blast crisis results in dismal treatment outcomes, and it is often fatal even with intervention [7]. Patients in AP and BC are generally grouped together as advanced-phase CML patients [8].
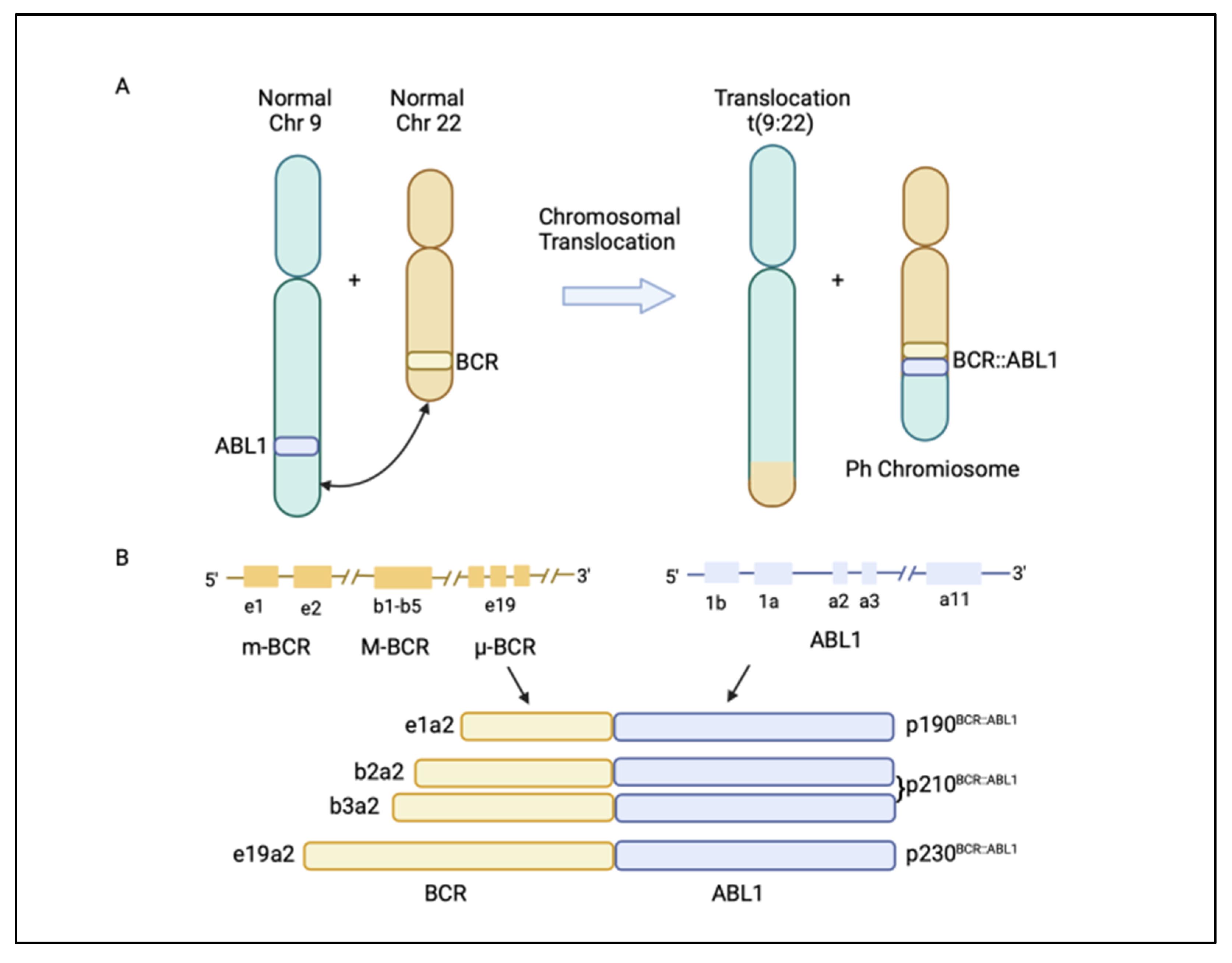
Figure 1. (A) Philadelphia (Ph) chromosome is formed from the translocation t(9;22)(q34;q11) of chromosome 9 and 22. The translocation event leads to the fusion of the breakpoint cluster region (BCR) gene with the Abelson1 proto-oncogene 1 (ABL1) gene, resulting in a BCR::ABL1 fusion gene. (B) Different BCR::ABL1 fusion gene transcripts p190BCR::ABL1, p210BCR::ABL1, and p230BCR::ABL1 are generated, depending on where the break occurs in the BCR gene [5]. Figure created in BioRender.com.
The BCR::ABL1 fusion gene is translated into the BCR::ABL1 protein, which contains several domains from BCR and ABL1. The BCR region of this protein regulates its enzymatic activity and provides sites for its binding partners [9][10]. The coil–coil domain of the BCR N-terminal part is responsible for the oligomerization and constitutive activation of BCR::ABL1 activity (Figure 2C) [11]. The ABL1 component of the BCR::ABL1 protein contains an SRC-homology-2 (SH2) domain, an SH3 domain, and a kinase domain [4]. When not fused with BCR, ABL1 has a myristoylated N-terminal responsible for the auto-inactivation of ABL1 kinase activity [4]. However, the myristoylated N-terminal is lost during the BCR::ABL1 fusion process [12]. The BCR:ABL1 kinase domain consists of key motifs responsible for its activity, including the phosphate binding loop (p-loop), the contact site (ATP/IM binding site), the catalytic domain, and activation loop (A-loop) (Figure 2C) [13]. BCR::ABL1 is active when ATP binds to the active site in the ABL1 kinase domain and transfers its phosphate group to ABL1 substrate. However, tyrosine kinase inhibitors (TKIs) compete with ATP for binding to the active site, inhibiting the BCR::ABL1 activation and preventing leukaemia (Figure 2A) [14].
The treatment of CML with TKIs has been paradigm-shifting, increasing the survival from 20% to over 80% today [14]. There are five available approved TKIs, prescribed based on disease phase, individual risk assessment, response level, presence of BCR::ABL1 kinase domain mutations, and response to prior TKI therapy [15][16]. The current available TKIs include the first-generation TKI imatinib (Glivec, Novartis, Basel, Switzerland ); second-generation TKIs dasatinib (Sprycel, Bristol-Myers Squibb, New York, USA), nilotinib (Tasigna, Novartis), and bosutinib (Bosulif, Pfizer, New York, USA); and third-generation ponatinib (Iclusig, Takeda/Incyte, Tokyo, Japan). Ponatinib is approved as a third-line treatment, when two or more TKIs are not effective, and for patients harbouring the T315I mutation in the BCR::ABL1 kinase domain. In October 2021, the Food and Drug Administration approved the new allosteric inhibitor of BCR::ABL1, asciminib (Scemblix, Novartis), for CML patients previously treated with two or more TKIs and for T315I mutations [17]. Asciminib binds to the myristoyl’s site on BCR::ABL1 and allosterically inhibits its activation, including that of BCR::ABL1 with T315I mutation, with very high selectivity, preventing downstream signalling [18]. ABL1 can normally self-regulate its activity via its engagement with the myristoylated N-terminal; however, in patients with CML, this ability is lost when ABL1 is fused with BCR (Figure 2B) [12]. Therefore, the binding of asciminib to a myristoyl pocket facilitates the inhibition of BCR::ABL1 activity by restoring its allosteric inhibition ability (Figure 2B).
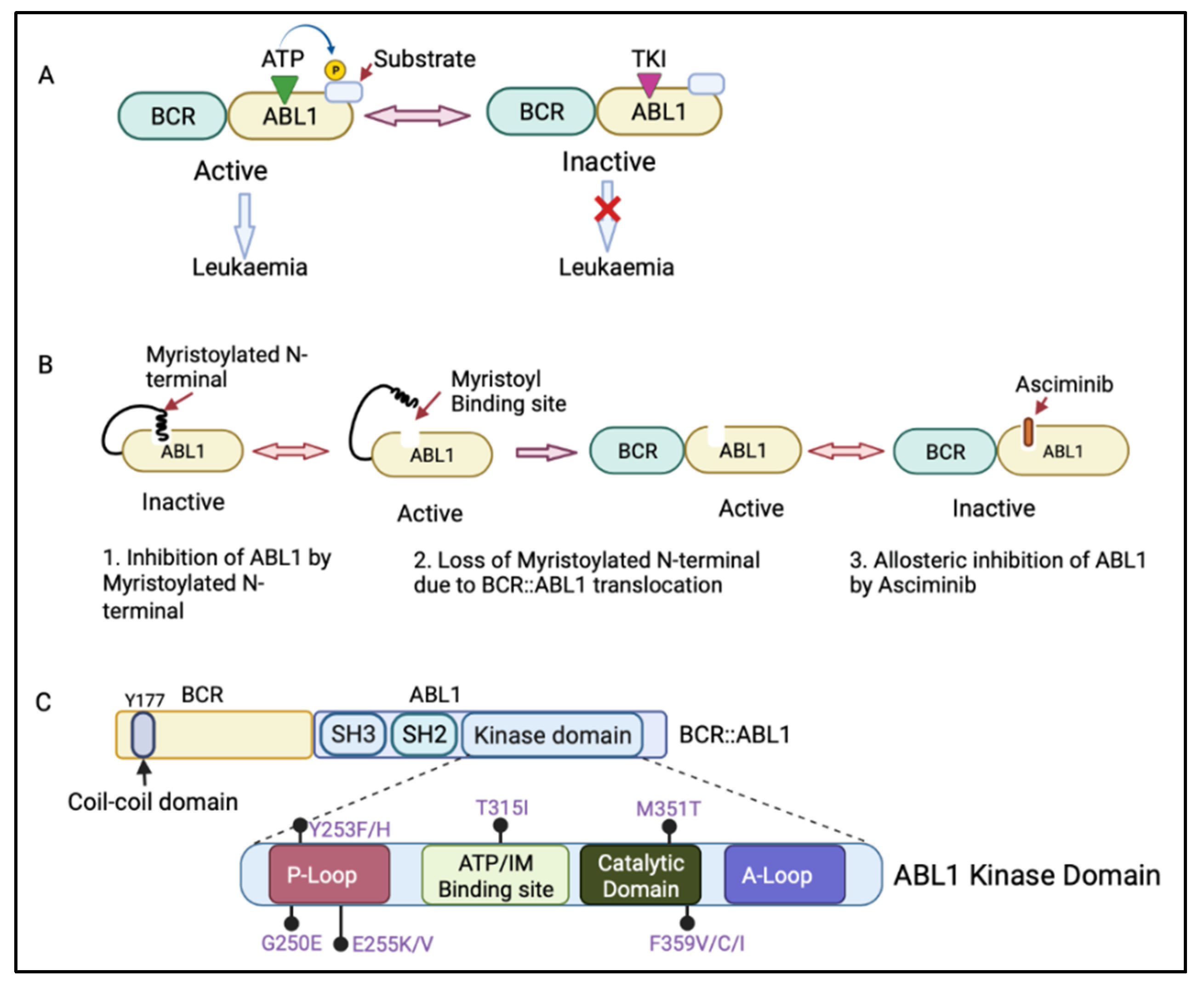
Figure 2. (A) Mechanism of action of adenosine triphosphate (ATP)-competitive tyrosine kinase inhibitors (TKIs). ATP binds to the ABL1 kinase domain, and the phosphate group is transferred to the ABL1 substrate, leading to BCR::ABL1 activation. The TKI competes with ATP for binding to the ABL1 kinase domain, inhibiting BCR::ABL1 activation and therefore preventing leukaemia progression. (B) Mechanism of action of asciminib, an allosteric inhibitor of BCR::ABL1. Asciminib binds to the myristoyl binding site, leading to BCR::ABL1 inactivation via allosteric inhibition of ABL1 kinase. (C) Schematic diagram of the BCR and ABL1 components of the BCR::ABL1 protein showing the N-terminal coil–coil domain (containing key tyrosine residue at 177 position, Y177) of BCR and an SRC-homology-2 (SH2) domain, an SH3 domain, and a kinase domain of ABL1. The ABL1 kinase domain shows the P-loop, ATP/imatinib binding site, catalytic domain, A-loop, and the most clinically relevant mutations affecting the kinase domain [19]. Figure created in BioRender.com.
The response to TKIs is assessed by measuring the levels of BCR::ABL1 transcript in the peripheral blood by quantitative real-time PCR (RT-qPCR) and based on the achievement of molecular milestones over time [20]. BCR::ABL1% is expressed and reported on a log scale, where 10%, 1%, 0.1%, 0.01%, 0.0032%, and 0.001% corresponds to 1, 2, 3, 4, 4.5, and 5 log reductions, respectively, below the standard baseline used in IRIS study [21]. Both 4-log (MR4) and 4.5-log (MR4.5) reductions are described as deep molecular responses [22]. Optimal molecular response corresponds to achieving specific milestones, which are the early molecular response of BCR::ABL1 ≤ 10% at 3 months and the major molecular response (MMR) to BCR::ABL1 ≤ 0.1% at 12 months. The main goal for CML patients is to achieve a durable remission, known as treatment-free remission, which first requires maintaining a deep molecular response (DMR) (BCR::ABL1 ≤ 0.01% or undetectable; limit of detection of 0.001%) [23]. Patients who achieve a deep molecular response (MR4 or MR4.5) have a better outcome with low risk of disease progression or relapse [22].
Chronic-phase CML patients who achieve an optimal response to treatment can expect a comparable life expectancy to that of the general population. About 25% of patients can cease their TKI therapy and maintain treatment-free remission [24][25][26][27]. Despite this enormous success, several challenges remain. The failure to eradicate persistent CML cells leads to a relapse in about 50% of patients who cease therapy. These patients therefore need to restart therapy, which can cause considerable emotional stress. Additionally, about 20% of CML patients respond poorly to frontline therapy, with 5–10% progressing to blast crisis [28]. This is due to the development of resistance, which represents a “bottleneck to cure” [29]. While a lot of progress has been made in understanding the BCR::ABL1-dependent mechanisms of resistance that rely on BCR::ABL1 reactivation, the mechanisms of BCR::ABL1-independent resistance have remained largely elusive [30]. It is becoming increasingly clear that TKI resistance can be driven by mechanisms that do not depend on BCR::ABL1 activation [31]. These mechanisms need to be considered in the curative approach of CML.
2. Targeted Therapies against BCR::ABL1-Independent Resistant Cells
Given the variety of BCR::ABL1-independent mechanisms of resistance, it is now important to focus the studies on the development of the most appropriate targeted therapy for a specific deregulated pathway. This will also involve the development of sensitive techniques for identifying specific biomarkers using phospho-flow and next-generation sequencing at diagnosis or at relapse [32]. Mutations in several genes have been identified in CML patients, such as ASXL1, RUNX1, TET2, BCL6 Corepressor-Like 1 (BCORL1), GATA-binding factor 2 (GATA2), and others [33]. Kim et al. showed a strong correlation between the mutations in TP53, Lysine Methyltransferase 2D (KMT2D), and TET2 during TKI therapy and treatment failure in CML patients [34]. Additionally, IKAROS Family Zinc Finger 1 (IKZF1) exon deletion has been reported to be associated with poorer outcomes in CML patients [33]. In an interesting study, Chan et al. discovered that mutations converging in one principal driver pathway were able to promote progression toward leukaemia [35]. Conversely, divergent signalling pathways represented a powerful barrier to transformations. Mutations in these divergent pathways prevented leukaemia instead of promoting it [35]. This finding is of principal relevance for the development of new targeted therapies against deregulated pathways in leukaemia. Chan et al. also demonstrated that targeting divergent pathways had a counterproductive effect on cancer progression. It is therefore vital to identify the driver pathway and target it [35]. This supports the possibility of developing drugs that could target different mediators of a driver pathway, increasing the possibility of finding the best precision medicine therapeutic approach.
In the context of BCR::ABL1-independent pathways, combination therapies that target both BCR::ABL1 and alternative survival pathways have the potential to eliminate leukaemic stem cells and sensitize progenitor cells, improving the treatment of CML. For instance, using a CML-mouse model, Shan et al. demonstrated that the inhibition of BCR::ABL1 with TKIs, combined with inhibition of the RAS pathway using trametinib, an FDA-approved MEK inhibitor, synergistically induced cell death in BCR::ABL1-independent MAPK pathway-driven imatinib-resistant CML stem cells [36]. Similarly, the JAK/STAT pathway has also been implicated in the survival of quiescent leukaemic stem cells, and the combination therapy of TKIs and ruxolitinib (JAK2 inhibitor) has been demonstrated to reduce their number in murine xenografts [37]. In a phase I clinical trial (NCT01702064), the combination therapy of nilotinib and ruxolitinib resulted in undetectable BCR::ABL1 in 40% of CML patients after 6 months, as measured by RT-qPCR [38]. As a result, this combination treatment was recommended for further investigation in a phase II clinical trial [38]. Moreover, Yagi et al. reported that the specific activation of STAT3 in CML and the combined inhibition of JAK1 with tofacitinib and BCR::ABL1 with imatinib synergistically induced anti-tumour effects in CML cells [39]. Likewise, many PI3K/AKT/mTOR pathway inhibitors, including those which have received FDA approval, such as PI3K inhibitors (Idelalisib, Copanlisib, and Duvelisib) and mTOR inhibitors (Everolimus, Sirolimus, and Temsirolimus), have been studied as potential treatments against TKI-resistant CML, and active pre-clinical investigation is ongoing (Figure 3 and Table 1) [40]. A clinical trial is underway for investigating the safety and efficacy of the co-treatment of imatinib and everolimus in CML (NCT00093639).
There are multiple clinical trials assessing the inhibitors of the WNT/β-catenin pathway in various haematological malignancies, including CML [41]. The results from phase I and II clinical trials are currently being evaluated for the safety and efficacy of PRI-724, an inhibitor of WNT/β-catenin pathway, in pancreatic cancer and leukaemia patients [41]. More recently, a combination treatment with dasatinib and okadaic acid (an inhibitor of PP2A) has also been shown to induce cell cycle arrest and apoptosis in the K562 cell line [42]. However, conflicting reports of the pro- and anti-survival role of PP2A warrants further investigation to understand the situations where such a dual role of PP2A exists [43]. Consistent with the previously reported tumour-suppressor role of PP2A, its activating drugs, such as FTY720 and OP449, have been shown to re-sensitize TKI-resistant LSCs to BCR::ABL1 inhibition by targeting the JAK2/PP2A/β-catenin pathway (Figure 3 and Table 1) [44]. Therefore, the combination of TKI and an FDA-approved PP2A activator FTY720 or OP449 has been actively investigated in preclinical studies [44].
Due to the ability of EZH2 to reprogram and prevent apoptosis in CML-leukaemic stem cells, the combination of TKI treatment and an FDA-approved inhibitor of EZH2, Tazemetostat, has also been actively investigated in pre-clinical trials [45]. Similarly, Stobo et al. demonstrated that the first-generation autophagy inhibitor hydroxychloroquine potentiated TKI-induced cell death in CML-leukaemic stem cells, but their recent clinical trial combining hydroxychloroquine with imatinib showed that clinically achievable doses of hydroxychloroquine were not sufficient to accomplish the efficient inhibition of autophagy [46]. Autophagy genes were found to be highly expressed in CML-patient-derived leukaemic stem cells compared with progenitor cells, and the genetic or pharmacological inhibition of autophagy with the second-generation autophagy inhibitor Lys05 resulted in decreased leukaemic cell viability and improved sensitivity to chemotherapy [47]. Therefore, the second-generation FDA-approved autophagy inhibitors Lys05 and PIK-III were combined with TKIs to investigate their effects in preclinical trials (Figure 3) [47].
There are ongoing clinical trials investigating the combination of TKIs and IFN-α to eliminate leukaemic stem cells, since IFN-α can augment immune responses while TKIs inhibit BCR::ABL1 to exert combined effects [48]. Palandri et al. revealed higher complete cytogenetic responses in the imatinib and IFN-α group compared with the imatinib-only group (60% vs. 42%, p = 0.003) at 6 months, with a more rapid response rate seen with the combination treatment [49]. However, there was no difference in the complete cytogenetic response rate at 48 months (88% vs. 88%). Another phase II study by the Nordic group showed higher major molecular response rates (82% vs. 54%) with an imatinib and IFN-α combination treatment compared with an imatinib-only treatment [50]. Another clinical trial investigating the combination of bosutinib with interferon is also underway, and results are yet to be published (NCT03831776) [51]. The combination therapy of TKIs and the hedgehog inhibitor trial was discontinued at a very early stage due to toxicity being observed [30]. If the drug could be further refined to minimize toxicity, there may be potential for using this combination treatment in CML.
Carter et al. first discovered that the anti-apoptotic protein Bcl-2 plays a key role in the survival of CML stem cells and progenitor cells, making it an attractive CML target [52]. The combined inhibition of Bcl-2 with venetoclax and TKI provided rationale for the potential curative treatment of CML (Figure 3 and Table 1) [52]. The phase II clinical trial (NCT02689440) combining TKI (dasatinib) and venetoclax in heavily pretreated blast-phase CML showed encouraging results, with a 75% overall response rate [53]. Another phase II clinical trial combining ponatinib, venetoclax, and decitabine in blast-phase CML is also underway, investigating the overall patient response rate (NCT04188405) [53].
Table 1. This table shows the BCR::ABL1-independent pathways and their inhibitors that could be used in combination with TKIs, and the stage of their current development for diseases listed that could be repurposed in the treatment of Ph+ leukaemias.
| Target Pathway | Inhibitor/s | Stage of Development |
Approved/Treated Disease | Ref. |
|---|---|---|---|---|
| RAS/RAF/MEK/ERK |
|
FDA approved | BRAF(V600E) melanoma | [54] |
|
FDA approved | |||
| JAK/STAT |
|
FDA approved |
|
[55][56] |
|
FDA approved |
|
||
|
FDA approved |
|
||
| PI3K/AKT/mTOR |
|
FDA approved |
|
[57][58] |
|
FDA approved |
|
||
|
FDA approved |
|
||
| Wnt/β-catenin | CBP/β-catenin antagonist: PRI-724 | Phase 2 Clinical Trial (NCT01606579) | Acute myeloid leukaemia and chronic myeloid leukaemia | [59] |
| Tumour suppressor: PP2A | SET: FTY720 (Fingolimod) | FDA Approved | Multiple myeloma and mantle cell lymphoma | [60][61] |
| Epigenetic modulator: EZH2 | Tazemetostat | FDA Approved | Advanced or metastatic epithelioid sarcoma | [62] |
| Immune system | IFN-α | FDA Approved | Hairy cell leukaemia, CML, follicular non-Hodgkin lymphoma, melanoma, and AIDS-related Kaposi’s sarcoma |
[63] |
| Hedgehog pathway | Vismodegib (GDC-0449) and Sonidegib (LDE225) | FDA Approved | Basal cell carcinoma and acute myeloid leukaemia |
[64] |
| Intrinsic apoptotic pathway | Venetoclax | FDA Approved | Chronic lymphocytic leukaemia and acute myeloid leukaemia |
[65] |
This entry is adapted from the peer-reviewed paper 10.3390/cancers14143300
References
- Bennett, J.H. Case of hypertrophy of the spleen and liver, in which death took place from suppuration of the blood. Edinburgh Med. Sug. J. 1845, 64, 413–423.
- Nowell, P.C.; Hungerford, D.A. Chromosome studies on normal and leukemic human leukocytes. J. Natl. Cancer Inst. 1960, 25, 85–109.
- Propp, S.; Lizzi, F.A. Brief Report: Philadelphia Chromosome in Acute Lymphocytic Leukemia. Blood 1970, 36, 353–360.
- Quintas-Cardama, A.; Cortes, J. Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood 2009, 113, 1619–1630.
- Flis, S.; Chojnacki, T. Chronic myelogenous leukemia, a still unsolved problem: Pitfalls and new therapeutic possibilities. Drug Des. Dev. Ther. 2019, 13, 825–843.
- Deininger, M.W.; Goldman, J.M.; Melo, J.V. The molecular biology of chronic myeloid leukemia. Blood 2000, 96, 3343–3356.
- Saxena, K.; Jabbour, E.; Issa, G.; Sasaki, K.; Ravandi, F.; Maiti, A.; Daver, N.; Kadia, T.; DiNardo, C.D.; Konopleva, M.; et al. Impact of frontline treatment approach on outcomes of myeloid blast phase CML. J. Hematol. Oncol. 2021, 14, 94.
- Sessions, J. Chronic myeloid leukemia in 2007. Am. J. Health-Syst. Pharm. 2007, 64, S4–S9.
- McWhirter, J.R.; Wang, J.Y. An actin-binding function contributes to transformation by the Bcr-Abl oncoprotein of Philadelphia chromosome-positive human leukemias. EMBO J. 1993, 12, 1533–1546.
- Peiris, M.N.; Li, F.; Donoghue, D.J. BCR: A promiscuous fusion partner in hematopoietic disorders. Oncotarget 2019, 10, 2738–2754.
- Pendergast, A.M.; Quilliam, L.A.; Cripe, L.D.; Bassing, C.H.; Dai, Z.; Li, N.; Batzer, A.; Rabun, K.M.; Der, C.J.; Schlessinger, J. BCR-ABL-induced oncogenesis is mediated by direct interaction with the SH2 domain of the GRB-2 adaptor protein. Cell 1993, 75, 175–185.
- Hantschel, O.; Nagar, B.; Guettler, S.; Kretzschmar, J.; Dorey, K.; Kuriyan, J.; Superti-Furga, G. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 2003, 112, 845–857.
- Vinhas, R.; Lourenco, A.; Santos, S.; Lemos, M.; Ribeiro, P.; de Sousa, A.B.; Baptista, P.V.; Fernandes, A.R. A novel BCR-ABL1 mutation in a patient with Philadelphia chromosome-positive B-cell acute lymphoblastic leukemia. Oncol. Targets Ther. 2018, 11, 8589–8598.
- Shawver, L.K.; Slamon, D.; Ullrich, A. Smart drugs: Tyrosine kinase inhibitors in cancer therapy. Cancer Cell 2002, 1, 117–123.
- Radich, J.P.; Deininger, M.; Abboud, C.N.; Altman, J.K.; Berman, E.; Bhatia, R.; Bhatnagar, B.; Curtin, P.; DeAngelo, D.J.; Gotlib, J.; et al. Chronic Myeloid Leukemia, Version 1.2019, NCCN Clinical Practice Guidelines in Oncology. J. Natl. Compr. Cancer Netw. 2018, 16, 1108–1135.
- Hughes, T.P.; Mauro, M.J.; Cortes, J.E.; Minami, H.; Rea, D.; DeAngelo, D.J.; Breccia, M.; Goh, Y.-T.; Talpaz, M.; Hochhaus, A.; et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. N. Engl. J. Med. 2019, 381, 2315–2326.
- FDA. FDA Approves Asciminib for Philadelphia Chromosome-Positive Chronic Myeloid Leukemia. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-asciminib-philadelphia-chromosome-positive-chronic-myeloid-leukemia (accessed on 9 December 2021).
- Wylie, A.A.; Schoepfer, J.; Jahnke, W.; Cowan-Jacob, S.W.; Loo, A.; Furet, P.; Marzinzik, A.L.; Pelle, X.; Donovan, J.; Zhu, W.; et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017, 543, 733–737.
- Soverini, S.; Mancini, M.; Bavaro, L.; Cavo, M.; Martinelli, G. Chronic myeloid leukemia: The paradigm of targeting oncogenic tyrosine kinase signaling and counteracting resistance for successful cancer therapy. Mol. Cancer 2018, 17, 49.
- Branford, S.; Fletcher, L.; Cross, N.C.; Muller, M.C.; Hochhaus, A.; Kim, D.W.; Radich, J.P.; Saglio, G.; Pane, F.; Kamel-Reid, S.; et al. Desirable performance characteristics for BCR-ABL measurement on an international reporting scale to allow consistent interpretation of individual patient response and comparison of response rates between clinical trials. Blood 2008, 112, 3330–3338.
- Baccarani, M.; Deininger, M.W.; Rosti, G.; Hochhaus, A.; Soverini, S.; Apperley, J.F.; Cervantes, F.; Clark, R.E.; Cortes, J.E.; Guilhot, F.; et al. European LeukemiaNet recommendations for the management of chronic myeloid leukemia: 2013. Blood 2013, 122, 872–884.
- Breccia, M.; Molica, M.; Colafigli, G.; Massaro, F.; Quattrocchi, L.; Latagliata, R.; Mancini, M.; Diverio, D.; Guarini, A.; Alimena, G.; et al. Prognostic factors associated with a stable MR4.5 achievement in chronic myeloid leukemia patients treated with imatinib. Oncotarget 2018, 9, 7534–7540.
- Wang, R.; Cong, Y.; Li, C.; Zhang, C.; Lin, H. Predictive value of early molecular response for deep molecular response in chronic phase of chronic myeloid leukemia. Medicine 2019, 98, e15222.
- Hughes, T.; Boquimpani, C.; Takahashi, N.; Benyamini, N.; Clementino, N.; Shuvaev, V.; Ailawadhi, S.; Lipton, J.; Turkina, A.; Paz, R. Durable treatment-free remission after stopping second line nilotinib in patients with chronic myeloid leukemia in chronic phase: ENESTOP 96-wk update. Haematologica 2017, 102, 75.
- Ross, D.M.; Pagani, I.S.; Shanmuganathan, N.; Kok, C.H.; Seymour, J.F.; Mills, A.K.; Filshie, R.J.; Arthur, C.K.; Dang, P.; Saunders, V.A.; et al. Long-term treatment-free remission of chronic myeloid leukemia with falling levels of residual leukemic cells. Leukemia 2018, 32, 2572–2579.
- Ross, D.M.; Hughes, T.P. Treatment-free remission in patients with chronic myeloid leukaemia. Nat. Rev. Clin. Oncol. 2020, 17, 493–503.
- Mahon, F.-X.; Réa, D.; Guilhot, J.; Guilhot, F.; Huguet, F.; Nicolini, F.; Legros, L.; Charbonnier, A.; Guerci, A.; Varet, B. Discontinuation of imatinib in patients with chronic myeloid leukaemia who have maintained complete molecular remission for at least 2 years: The prospective, multicentre Stop Imatinib (STIM) trial. Lancet Oncol. 2010, 11, 1029–1035.
- Bonifacio, M.; Stagno, F.; Scaffidi, L.; Krampera, M.; Di Raimondo, F. Management of Chronic Myeloid Leukemia in Advanced Phase. Front. Oncol 2019, 9, 1132.
- Soverini, S.; Bassan, R.; Lion, T. Treatment and monitoring of Philadelphia chromosome-positive leukemia patients: Recent advances and remaining challenges. J. Hematol. Oncol. 2019, 12, 39.
- Braun, T.P.; Eide, C.A.; Druker, B.J. Response and resistance to BCR-ABL1-targeted therapies. Cancer Cell 2020, 37, 530–542.
- Loscocco, F.; Visani, G.; Galimberti, S.; Curti, A.; Isidori, A. BCR-ABL Independent Mechanisms of Resistance in Chronic Myeloid Leukemia. Front. Oncol. 2019, 9, 939.
- Medina, A.; Puig, N.; Flores-Montero, J.; Jimenez, C.; Sarasquete, M.E.; Garcia-Alvarez, M.; Prieto-Conde, I.; Chillon, C.; Alcoceba, M.; Gutierrez, N.C.; et al. Comparison of next-generation sequencing (NGS) and next-generation flow (NGF) for minimal residual disease (MRD) assessment in multiple myeloma. Blood Cancer J. 2020, 10, 108.
- Branford, S.; Kim, D.D.H.; Apperley, J.F.; Eide, C.A.; Mustjoki, S.; Ong, S.T.; Nteliopoulos, G.; Ernst, T.; Chuah, C.; Gambacorti-Passerini, C.; et al. Laying the foundation for genomically-based risk assessment in chronic myeloid leukemia. Leukemia 2019, 33, 1835–1850.
- Kim, T.; Tyndel, M.S.; Kim, H.J.; Ahn, J.S.; Choi, S.H.; Park, H.J.; Kim, Y.K.; Kim, S.Y.; Lipton, J.H.; Zhang, Z.; et al. Spectrum of somatic mutation dynamics in chronic myeloid leukemia following tyrosine kinase inhibitor therapy. Blood 2017, 129, 38–47.
- Chan, L.N.; Murakami, M.A.; Robinson, M.E.; Caeser, R.; Sadras, T.; Lee, J.; Cosgun, K.N.; Kume, K.; Khairnar, V.; Xiao, G.; et al. Signalling input from divergent pathways subverts B cell transformation. Nature 2020, 583, 845–851.
- Ma, L.; Shan, Y.; Bai, R.; Xue, L.; Eide, C.A.; Ou, J.; Zhu, L.J.; Hutchinson, L.; Cerny, J.; Khoury, H.J.; et al. A therapeutically targetable mechanism of BCR-ABL-independent imatinib resistance in chronic myeloid leukemia. Sci. Transl. Med. 2014, 6, 252ra121.
- Gallipoli, P.; Cook, A.; Rhodes, S.; Hopcroft, L.; Wheadon, H.; Whetton, A.D.; Jorgensen, H.G.; Bhatia, R.; Holyoake, T.L. JAK2/STAT5 inhibition by nilotinib with ruxolitinib contributes to the elimination of CML CD34+ cells in vitro and in vivo. Blood 2014, 124, 1492–1501.
- Sweet, K.; Hazlehurst, L.; Sahakian, E.; Powers, J.; Nodzon, L.; Kayali, F.; Hyland, K.; Nelson, A.; Pinilla-Ibarz, J. A phase I clinical trial of ruxolitinib in combination with nilotinib in chronic myeloid leukemia patients with molecular evidence of disease. Leuk. Res. 2018, 74, 89–96.
- Yagi, K.; Shimada, A.; Sendo, T. Pharmacological inhibition of JAK3 enhances the antitumor activity of imatinib in human chronic myeloid leukemia. Eur. J. Pharmacol. 2018, 825, 28–33.
- Singh, P.; Kumar, V.; Gupta, S.K.; Kumari, G.; Verma, M. Combating TKI resistance in CML by inhibiting the PI3K/Akt/mTOR pathway in combination with TKIs: A review. Med. Oncol. 2021, 38, 10.
- Zhang, Y.; Wang, X. Targeting the Wnt/beta-catenin signaling pathway in cancer. J. Hematol. Oncol. 2020, 13, 165.
- Ozel, B.; Kipcak, S.; Biray Avci, C.; Gunduz, C.; Saydam, G.; Aktan, C.; Selvi Gunel, N. Combination of dasatinib and okadaic acid induces apoptosis and cell cycle arrest by targeting protein phosphatase PP2A in chronic myeloid leukemia cells. Med. Oncol. 2022, 39, 46.
- Perrotti, D.; Agarwal, A.; Lucas, C.M.; Narla, G.; Neviani, P.; Odero, M.D.; Ruvolo, P.P.; Verrills, N.M. Comment on “PP2A inhibition sensitizes cancer stem cells to ABL tyrosine kinase inhibitors in BCR-ABL human leukemia”. Sci. Transl. Med. 2019, 11, eaau0416.
- Mazhar, S.; Taylor, S.E.; Sangodkar, J.; Narla, G. Targeting PP2A in cancer: Combination therapies. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 51–63.
- Scott, M.T.; Korfi, K.; Saffrey, P.; Hopcroft, L.E.M.; Kinstrie, R.; Pellicano, F.; Guenther, C.; Gallipoli, P.; Cruz, M.; Dunn, K.; et al. Epigenetic Reprogramming Sensitizes CML Stem Cells to Combined EZH2 and Tyrosine Kinase Inhibition. Cancer Discov. 2016, 6, 1248.
- Horne, G.A.; Stobo, J.; Kelly, C.; Mukhopadhyay, A.; Latif, A.L.; Dixon-Hughes, J.; McMahon, L.; Cony-Makhoul, P.; Byrne, J.; Smith, G.; et al. A randomised phase II trial of hydroxychloroquine and imatinib versus imatinib alone for patients with chronic myeloid leukaemia in major cytogenetic response with residual disease. Leukemia 2020, 34, 1775–1786.
- Baquero, P.; Dawson, A.; Mukhopadhyay, A.; Kuntz, E.M.; Mitchell, R.; Olivares, O.; Ianniciello, A.; Scott, M.T.; Dunn, K.; Nicastri, M.C. Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia 2019, 33, 981–994.
- Mu, H.; Zhu, X.; Jia, H.; Zhou, L.; Liu, H. Combination Therapies in Chronic Myeloid Leukemia for Potential Treatment-Free Remission: Focus on Leukemia Stem Cells and Immune Modulation. Front. Oncol. 2021, 11, 643382.
- Palandri, F.; Castagnetti, F.; Iacobucci, I.; Martinelli, G.; Amabile, M.; Gugliotta, G.; Poerio, A.; Testoni, N.; Breccia, M.; Bocchia, M.; et al. The response to imatinib and interferon-alpha is more rapid than the response to imatinib alone: A retrospective analysis of 495 Philadelphia-positive chronic myeloid leukemia patients in early chronic phase. Haematologica 2010, 95, 1415–1419.
- Simonsson, B.; Gedde-Dahl, T.; Markevarn, B.; Remes, K.; Stentoft, J.; Almqvist, A.; Bjoreman, M.; Flogegard, M.; Koskenvesa, P.; Lindblom, A.; et al. Combination of pegylated IFN-alpha2b with imatinib increases molecular response rates in patients with low- or intermediate-risk chronic myeloid leukemia. Blood 2011, 118, 3228–3235.
- Gisslinger, H.; Zagrijtschuk, O.; Buxhofer-Ausch, V.; Thaler, J.; Schloegl, E.; Gastl, G.A.; Wolf, D.; Kralovics, R.; Gisslinger, B.; Strecker, K.; et al. Ropeginterferon alfa-2b, a novel IFN.Nalpha-2b, induces high response rates with low toxicity in patients with polycythemia vera. Blood 2015, 126, 1762–1769.
- Carter, B.Z.; Mak, P.Y.; Mu, H.; Zhou, H.; Mak, D.H.; Schober, W.; Leverson, J.D.; Zhang, B.; Bhatia, R.; Huang, X. Combined targeting of BCL-2 and BCR-ABL tyrosine kinase eradicates chronic myeloid leukemia stem cells. Sci. Transl. Med. 2016, 8, ra117–ra355.
- Maiti, A.; Franquiz, M.J.; Ravandi, F.; Cortes, J.E.; Jabbour, E.J.; Sasaki, K.; Marx, K.; Daver, N.G.; Kadia, T.M.; Konopleva, M.Y.; et al. Venetoclax and BCR-ABL Tyrosine Kinase Inhibitor Combinations: Outcome in Patients with Philadelphia Chromosome-Positive Advanced Myeloid Leukemias. Acta Haematol. 2020, 143, 567–573.
- Samatar, A.A.; Poulikakos, P.I. Targeting RAS-ERK signalling in cancer: Promises and challenges. Nat. Rev. Drug Discov. 2014, 13, 928–942.
- Han, E.S.; Wen, W.; Dellinger, T.H.; Wu, J.; Lu, S.A.; Jove, R.; Yim, J.H. Ruxolitinib synergistically enhances the anti-tumor activity of paclitaxel in human ovarian cancer. Oncotarget 2018, 9, 24304–24319.
- Mogul, A.; Corsi, K.; McAuliffe, L. Baricitinib: The Second FDA-Approved JAK Inhibitor for the Treatment of Rheumatoid Arthritis. Ann. Pharmacother. 2019, 53, 947–953.
- Song, M.; Bode, A.M.; Dong, Z.; Lee, M.-H. AKT as a therapeutic target for cancer. Cancer Res. 2019, 79, 1019–1031.
- Blagosklonny, M.V. Rapamycin for longevity: Opinion article. Aging 2019, 11, 8048–8067.
- Jung, Y.S.; Park, J.I. Wnt signaling in cancer: Therapeutic targeting of Wnt signaling beyond beta-catenin and the destruction complex. Exp. Mol. Med. 2020, 52, 183–191.
- Tinsley, S.L.; Allen-Petersen, B.L. PP2A and cancer epigenetics: A therapeutic opportunity waiting to happen. NAR Cancer 2022, 4, zcac002.
- O’Connor, C.M.; Perl, A.; Leonard, D.; Sangodkar, J.; Narla, G. Therapeutic targeting of PP2A. Int. J. Biochem. Cell Biol. 2018, 96, 182–193.
- Li, C.; Wang, Y.; Gong, Y.; Zhang, T.; Huang, J.; Tan, Z.; Xue, L. Finding an easy way to harmonize: A review of advances in clinical research and combination strategies of EZH2 inhibitors. Clin. Epigenet. 2021, 13, 62.
- Borden, E.C. Interferons alpha and beta in cancer: Therapeutic opportunities from new insights. Nat. Rev. Drug Discov. 2019, 18, 219–234.
- Jamieson, C.; Martinelli, G.; Papayannidis, C.; Cortes, J.E. Hedgehog Pathway Inhibitors: A New Therapeutic Class for the Treatment of Acute Myeloid Leukemia. Blood Cancer Discov. 2020, 1, 134–145.
- Lasica, M.; Anderson, M.A. Review of Venetoclax.x in CLL, AML and Multiple Myeloma. J. Pers. Med. 2021, 11, 463.
This entry is offline, you can click here to edit this entry!