Your browser does not fully support modern features. Please upgrade for a smoother experience.
Please note this is an old version of this entry, which may differ significantly from the current revision.
Subjects:
Toxicology
Poly- and perfluoroalkylated substances (PFAS) are chemicals that persist and bioaccumulate in the environment and are found in nearly all human populations through several routes of exposure. Human occupational and community exposure to PFAS has been associated with several cancers, including cancers of the kidney, testis, prostate, and liver.
- epigenetics
- PFOA
- testicular cancer
1. Introducition
Unlike known carcinogens such as benzo(a)pyrene and UV light that are genotoxic due to direct damage to DNA, there is little evidence that PFAS are direct mutagens or deregulators of DNA repair or genomic stability [37,38,39]. However, at high concentrations, PFAS have been demonstrated to damage DNA via reactive oxygen species generation [40,41]. It is unclear if this mechanism is relevant for typical levels of human PFAS exposure. In contrast, most of the evidence for PFAS-mediated effects has focused on epigenetics, transcription, cellular metabolism, and endocrine effects [11,12,37,42,43,44].
2. Metabolism
Metabolic plasticity is one of the hallmarks of cancer [45]. PFAS exposure causes numerous metabolic alterations, through both PPAR-dependent and -independent mechanisms in the liver and other tissues [11,42]. Structurally, PFAS resemble fatty acids (FAs) and there is evidence that PFAS can act as ligands for peroxisome proliferator-activated receptors (PPARs) [46,47]. PPARs are transcription factors with many biological effects beyond their canonical role in controlling lipid and glucose metabolism [48]. Hence, activation of PPARs is an attractive mechanism to explain many of the biological effects of PFAS. The activation of PPARα has been extensively studied as a mechanism of PFAS-mediated liver toxicities, including fibrosis, cirrhosis, steatosis, non-alcoholic fatty acid liver disease, and liver cancer [49,50,51,52]. Similarly, the PFAS activation of PPARs has also been proposed to mediate dyslipidemia (especially high cholesterol), insulin resistance, adipogenesis, and several cancers, including colon, breast, and prostate cancer [11,42,53,54,55,56,57]. Likely related again to a structural similarity with FAs, PFAS are known to accumulate in the liver and have been proposed as altering FA metabolism by binding to FA transporters and metabolic enzymes [11,42]. In contrast to PFAS activation of PPARs, there is less evidence for direct activation by PFAS of other metabolic and xenobiotic nuclear receptors that respond to FAs, including liver X (LXR), farnesoid X (FXR), constitutive androstane (CAR), and pregnane X (PXR). Since altered metabolism is a key feature of the cancer phenotype, the alteration of metabolic regulators such as PPARs offers an attractive mechanism for the proposed pro-carcinogenetic actions of PFAS [45]. Another mechanism related to FA mimicry is the proposed direct effect of PFAS on regulating cell membrane fluidity [58,59]. Published studies demonstrate a central role for PPARα signaling in PFOA/PFOS-induced liver and kidney carcinogenesis [21,60]. In addition, an important role for fatty acid metabolism has been proposed for other cancers including breast, prostate, and colon cancer [61,62,63].
PFOA has been proposed to increase the risk of metabolic syndrome in humans [57]. PFAS alter the hepatic metabolism, with alterations in amino acid biogenesis and the Krebs cycle [64]. In addition, the upregulation of enzymes involved in β-oxidation has been reported upon PFOS exposure [65]. PFOS also induced high peroxisome, endoplasmic reticulum, mitochondria, and membrane protein levels, and deregulated lipid and amino acid metabolism [66,67]. Prenatal exposure to PFAS can contribute to pediatric liver toxicity [68]. A study of 1105 mother-child pairs that assessed multiple PFAS in maternal blood found higher liver enzyme levels of alanine aminotransferase, aspartate aminotransferase and gamma-glutamyl transferase [68]. Furthermore, PFAS levels were associated with alterations in serum amino acid levels in children [69]. In a study of male Chinese subjects, six PFAS were associated with metabolic serum changes associated with oxidative stress [70]. Metabolic stress, as evidenced by metabolites of oxidative DNA damage and lipid peroxidation, has also been documented for both animal and cell line studies for a number of PFAS compounds [54,70]. An additional study of targeted metabolomics found perturbations in branched-chain and aromatic amino acid biosynthesis and glycerophospholipid metabolism and a link between PFAS and increased risk of non-alcoholic steatohepatitis in children [68]. Rodent experiments have shown that early and prenatal PFAS is associated with liver injury in offspring [71,72].
In summary, the activation of PPARs and associated metabolic perturbations, especially in the liver, is one of the most studied mechanisms of PFAS actions. The recent appreciation that many cancers are driven and sustained by metabolic reprogramming underscores the potential importance of this pathway in studying the proposed pro-carcinogenic effects of PFAS. How metabolic reprogramming at the hepatic and cancer cell/cancer progenitor cell level cross-talks with epigenetic and endocrine reprogramming is a key area of future research for understanding the potential carcinogenicity of PFAS (Figure 1).
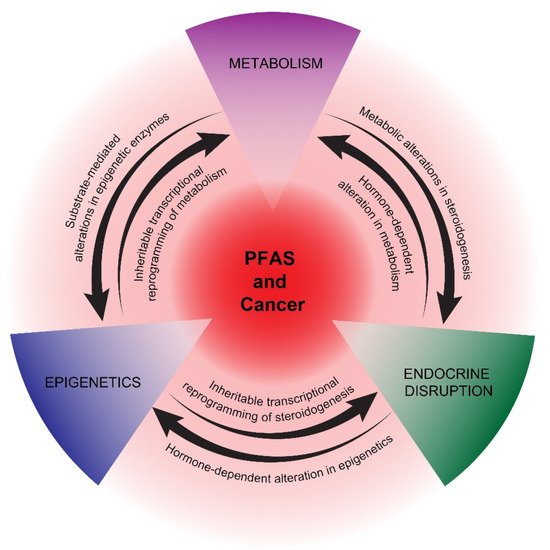
Figure 1. Proposed mechanisms of potential PFAS cancer promotion. PPAR-dependent and -independent reprogramming of metabolism, epigenetics, and endocrine disruption are represented as interconnecting, mutually reinforcing pathways of potential PFAS tumor promotion. The precise details of how PFAS influences these pathways are still uncertain, as is the impact of other proposed PFAS mechanisms, including immunosuppression and oxidative stress.
3. Endocrine Disruption
PFAS cross the placenta and concentrate in breast milk; thus, exposure to the developing fetus and infant occurs [73,74]. PFAS are known to have endocrine-disrupting properties [75,76]. There are reports of adverse reproductive health and decreased fecundity linked to PFAS exposure [77,78]. Human semen quality has decreased over the last several decades. This time period coincides with the rise in production of endocrine-disrupting chemicals (EDCs), and PFAS have been associated with infertility in male mice and subfertility in female mice [79,80]. In several studies, estrogenic and anti-androgen activities were observed for a number of PFAS compounds [81,82,83,84]. There is evidence that PFAS exposure is associated with decreased testosterone and poor sperm quality and numbers in humans [78,85]. For example, in a Japanese study, in utero PFOA and PFOS exposure was associated with decreased testosterone in male neonates [86]. In addition to in human studies, in rodents, PFAS have been observed to alter testosterone and estrogen levels, and were associated with impaired spermatogenesis and steroidogenesis and reduced sperm quality [81,82,83,84], although some inconsistent findings also exist [87]. In female rodents, PFOA alters mammary development [88,89]. PFOA has been associated with changes in the uterus and the reproductive health of female mice [90].
Several cancers associated with PFAS are hormone-dependent, including prostate and breast cancer, or have an etiology closely associated with endocrine disruption, as in testicular cancer [22,23,24,25,26,27,28]. In addition, endometrial cancer has been associated with endocrine disruption [91]. There is evidence that PFAS can alter endocrine hormone levels, potentially leading to disrupted reproductive health, especially with neonatal or pubertal exposure [92,93,94]. A major proposed mechanism of EDCs, in general, is their binding to nuclear receptors [95]. While there is strong evidence supporting the direct activation of PPARs, there is less evidence that PFAS directly activate endocrine receptors, including estrogen (ER) and androgen receptors (AR). Hence, the mechanism of endocrine disruption mediated by PFAS remains unclear, suggesting that indirect mechanisms, including epigenetic and/or metabolic reprogramming, may play roles in disrupting the production and secretion of endocrine hormones during critical windows of exposure [44,96] (Figure 1). In turn, early-life exposure to EDCs has been associated with epigenetic reprogramming that manifests later in life [97].
4. Epigenetics
Despite the likelihood that non-mutagenic, epigenetic pathways play a major role in PFAS biological effects, studies have been sparse, and these have mainly focused on DNA methylation. PFAS have been shown to be associated with both hypomethylation and hypermethylation in genome-wide and gene-specific molecular epidemiology studies [98,99,100,101,102,103,104,105]. PFAS levels have also been linked to decreased and differential DNA methylation in infants [102,103,104]. For example, reduced insulin-like growth factor methylation in cord blood was observed with prenatal PFOA exposure [104]. Another study reported that PFAS exposure was associated with increased long interspersed nuclear element-1 methylation [99]. Associations between PFAS exposures and altered methylation, either genome-wide or at specific loci, have been described in limited in vitro and animal studies, including early life exposures in rodents [106,107,108,109,110,111,112,113,114,115,116]. One study revealed PFOA-mediated hypomethylation of the glutathione-S-transferase Pi gene in liver cells [108]. Significant alterations in DNA methylation have been reported in vitro in HepG2 cells and in vivo in mouse kidney and liver tissues [111,112,113]. Globally, DNA methylation is altered during PFOS-induced fat cell differentiation [109]. Additionally, recent studies have reported PFAS-mediated alterations of epigenetic regulators, such as DNA methyltransferases, ten-eleven translocation methylcytosine dioxygenases, and histone deacetylase enzymes in different mouse organs and human cell lines [106,107,110,117,118,119]. PFAS-mediated effects on histone modifications and microRNAs have also been described [49,106,107,118,120,121,122].
In summary, epigenetics may play a key role in initiating and maintaining potential pro-cancerous states mediated by non-mutagenic PFAS chemicals. Despite this, very few mechanistic studies have been reported. We speculate that epigenetic reprogramming by PFAS may be driven, in part, by metabolomic alterations in substrates and cofactors of epigenetic enzymes and, reciprocally, that epigenetic-mediated, transcriptional reprogramming plays a key role in establishing and stabilizing the metabolic and hormonal states required for continued tumorigenesis [123,124,125,126,127] (Figure 1). This hypothesis is motivated by the above-mentioned association between PFAS and metabolic, epigenetic, and endocrine disruptions and the recent appreciation of mechanistic relationships between these three pathways. In the following section, we highlight these principles with two cancers possessing epidemiologic links to PFAS: prostate cancer, which is strongly associated with metabolic disruption, and testicular cancer, which is strongly associated with epigenetic reprogramming.
This entry is adapted from the peer-reviewed paper 10.3390/cancers14122919
This entry is offline, you can click here to edit this entry!