Alpha-synuclein (α-syn) is a small protein composed of 140 amino acids and belongs to the group of intrinsically disordered proteins. It is a soluble protein that is highly expressed in neurons and expressed at low levels in glial cells. The monomeric protein aggregation process induces the formation of oligomeric intermediates and proceeds towards fibrillar species. These α-syn conformational species have been detected in the extracellular space and mediate consequences on surrounding neurons and glial cells. In particular, higher-ordered α-syn aggregates are involved in microglial and oligodendrocyte activation, as well as in the induction of astrogliosis. These phenomena lead to mitochondrial dysfunction, reactive oxygen and nitrogen species formation, and the induction of an inflammatory response, associated with neuronal cell death. Several receptors participate in cell activation and/or in the uptake of α-syn, which can vary depending on the α-syn aggregated state and cell types. The receptors involved in this process are of outstanding relevance because they may constitute potential therapeutic targets for the treatment of PD and related synucleinopathies.
- extracellular alpha-synuclein
- oligomers
- fibrils
- astrocytes
- oligodendrocytes
- microglia
1. Introduction
1.1. Synucleinopathies
1.2. Alpha-Synuclein and Aggregation Process
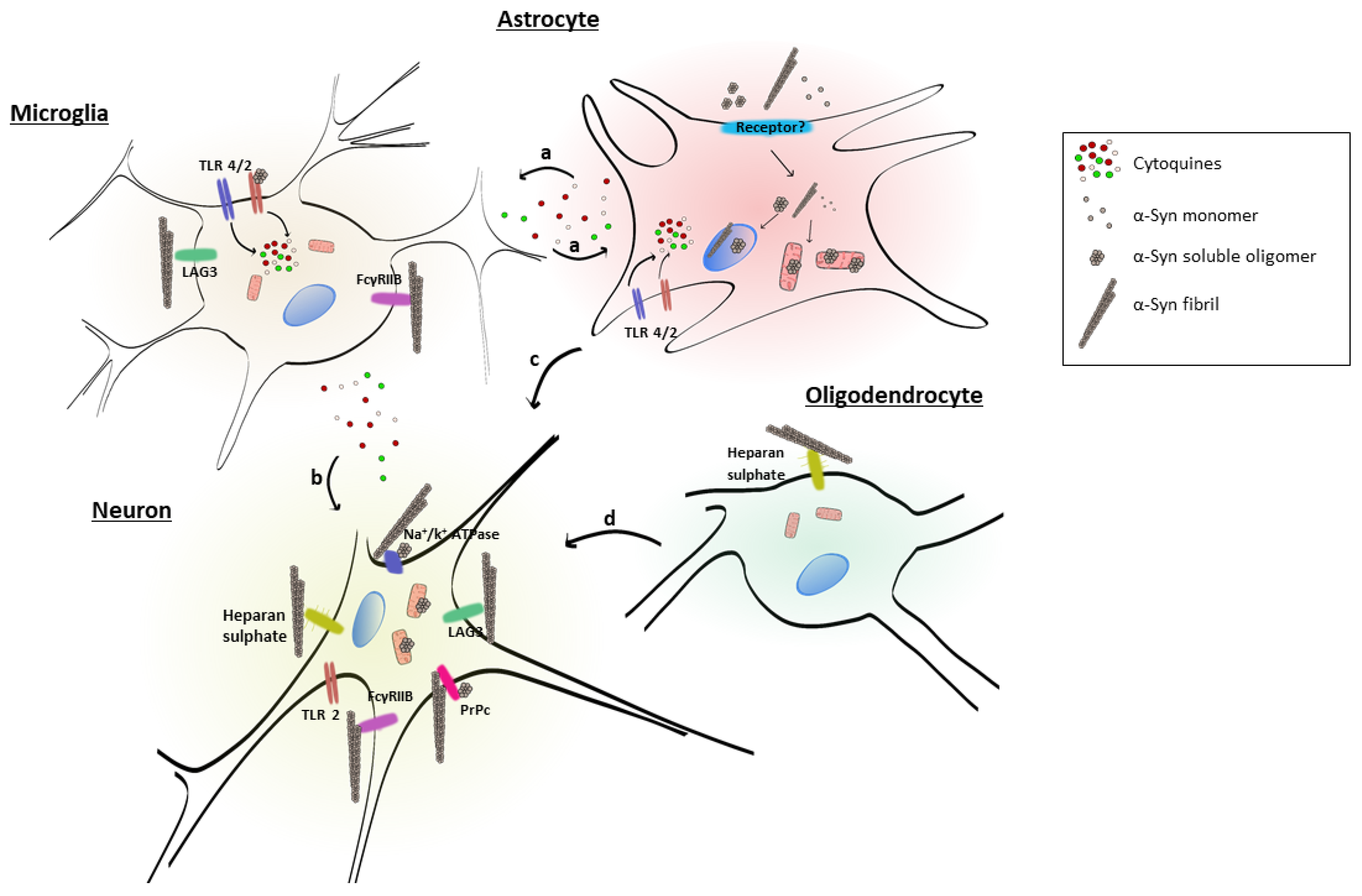
2. Extracellular α-Synuclein
2.1. Putative Mechanisms of α-Syn Uptake in Cells of the Nervous System
2.2. Glial Cell Uptake of Extracellular α-Syn and Activation
2.2.1. Role of Astrocytes
2.2.2. Role of Oligodendrocytes
2.2.3. Role of Microglia
2. Receptors for Extracellular α-Syn
| Receptor | α-Syn Conformer | Cell Type | Reference |
|---|---|---|---|
| Lymphocyte-activation gene 3 (LAG3) | Fibrils | Neuron, microglia | [102][103] |
| Cellular prion protein (PrPc) | Fibrils, soluble oligomers | Neurons | [104][105] |
| Heparan sulfate | Fibrils | Neuron, oligodendrocytes | [106] |
| Toll-like receptor 4 (TLR4) | n.d. | Microglia | [107] |
| Toll-like receptor 2 (TLR2) | Soluble oligomers | Microglia | [108] |
| a3-subunit of Na+/K+-ATPase | Fibrils, soluble oligomers | Neurons | [109] |
| FcƔRIIB | Fibrils | Microglia, neurons | [110] |
These data suggest that extracellular α-syn fibrils can bind to LAG3 and contribute to protein-induced dopaminergic neuronal loss and neurotoxicity [103]. This receptor may play a role in α-syn spreading pathology and neurodegeneration in PD and could be considered as a therapeutic target to avoid α-syn pathology.
This entry is adapted from the peer-reviewed paper 10.3390/biom12050655
References
- Parkinson, J. “An essay on the shaking palsy” 200 years old. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236.
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Prim. 2017, 3, 17013.
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066.
- Dauer, W.; Przedborski, S. Parkinson’s Disease: Mechanisms and Models. Camb. Companion Philos. Biol. 2003, 39, 889–909.
- Kalia, L.V.; Lang, A.E. Parkinson’s disease. Lancet 2015, 386, 896–912.
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884.
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Cairns, N.J.; Lantos, P.L.; Goedert, M. Filamentous alpha-synuclein inclusions link multiple system atrophy with Parkinson’s disease and dementia with Lewy bodies. Neurosci. Lett. 1998, 251, 205–208.
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173.
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841.
- Blesa, J.; Przedborski, S. Parkinson’s disease: Animal models and dopaminergic cell vulnerability. Front. Neuroanat. 2014, 8, 155.
- Dehay, B.; Fernagut, P.O. Alpha-synuclein-based models of Parkinson’s disease. Rev. Neurol. 2016, 172, 371–378.
- Duty, S.; Jenner, P. Animal models of Parkinson’s disease: A source of novel treatments and clues to the cause of the disease. Br. J. Pharmacol. 2011, 164, 1357–1391.
- Koprich, J.B.; Kalia, L.V.; Brotchie, J.M. Animal models of α-synucleinopathy for Parkinson disease drug development. Nat. Rev. Neurosci. 2017, 18, 515–529.
- Uchihara, T.; Giasson, B.I. Propagation of alpha-synuclein pathology: Hypotheses, discoveries, and yet unresolved questions from experimental and human brain studies. Acta Neuropathol. 2016, 131, 49–73.
- Schaser, A.J.; Stackhouse, T.L.; Weston, L.J.; Kerstein, P.C.; Osterberg, V.R.; López, C.S.; Dickson, D.W.; Luk, K.C.; Meshul, C.K.; Woltjer, R.L.; et al. Trans-synaptic and retrograde axonal spread of Lewy pathology following pre-formed fibril injection in an in vivo A53T alpha-synuclein mouse model of synucleinopathy. Acta Neuropathol. Commun. 2020, 8, 150.
- Lee, V.M.; Trojanowski, J.Q. Mechanisms of Parkinson’s disease linked to pathological alpha-synuclein: New targets for drug discovery. Neuron 2006, 52, 33–38.
- Savitt, J.M.; Dawson, V.L.; Dawson, T.M. Diagnosis and treatment of Parkinson disease: Molecules to medicine. J. Clin. Investig. 2006, 116, 1744–1754.
- Chandra, S.; Chen, X.; Rizo, J.; Jahn, R.; Sudhof, T.C. A broken alpha -helix in folded alpha -Synuclein. J. Biol. Chem. 2003, 278, 15313–15318.
- Davidson, W.S.; Jonas, A.; Clayton, D.F.; George, J.M. Stabilization of alpha-synuclein secondary structure upon binding to synthetic membranes. J. Biol. Chem. 1998, 273, 9443–9449.
- George, J.M. The synucleins. Genome Biol. 2002, 3, reviews3002.1.
- Kahle, P.J.; Haass, C.; Kretzschmar, H.A.; Neumann, M. Structure/function of alpha-synuclein in health and disease: Rational development of animal models for Parkinson’s and related diseases. J. Neurochem. 2002, 82, 449–457.
- Lavedan, C. The synuclein family. Genome Res. 1998, 8, 871–880.
- Culvenor, J.G.; McLean, C.A.; Cutt, S.; Campbell, B.C.; Maher, F.; Jakala, P.; Hartmann, T.; Beyreuther, K.; Masters, C.L.; Li, Q.X. Non-Abeta component of Alzheimer’s disease amyloid (NAC) revisited. NAC and alpha-synuclein are not associated with Abeta amyloid. Am. J. Pathol. 1999, 155, 1173–1181.
- el-Agnaf, O.M.; Irvine, G.B. Aggregation and neurotoxicity of alpha-synuclein and related peptides. Biochem. Soc. Trans. 2002, 30, 559–565.
- Ferrer, I. Alpha-synucleinopathies. Neurologia 2001, 16, 163–170.
- Ueda, K.; Fukushima, H.; Masliah, E.; Xia, Y.; Iwai, A.; Yoshimoto, M.; Otero, D.A.; Kondo, J.; Ihara, Y.; Saitoh, T. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 11282–11286.
- Hoyer, W.; Antony, T.; Cherny, D.; Heim, G.; Jovin, T.M.; Subramaniam, V. Dependence of alpha-synuclein aggregate morphology on solution conditions. J. Mol. Biol. 2002, 322, 383–393.
- Goedert, M.; Masuda-Suzukake, M.; Falcon, B. Like prions: The propagation of aggregated tau and α-synuclein in neurodegeneration. Brain 2017, 140, 266–278.
- Conway, K.A.; Harper, J.D.; Lansbury, P.T., Jr. Fibrils formed in vitro from alpha-synuclein and two mutant forms linked to Parkinson’s disease are typical amyloid. Biochemistry 2000, 39, 2552–2563.
- Kayed, R.; Sokolov, Y.; Edmonds, B.; McIntire, T.M.; Milton, S.C.; Hall, J.E.; Glabe, C.G. Permeabilization of lipid bilayers is a common conformation-dependent activity of soluble amyloid oligomers in protein misfolding diseases. J. Biol. Chem. 2004, 279, 46363–46366.
- Kim, H.Y.; Cho, M.K.; Kumar, A.; Maier, E.; Siebenhaar, C.; Becker, S.; Fernandez, C.O.; Lashuel, H.A.; Benz, R.; Lange, A.; et al. Structural properties of pore-forming oligomers of alpha-synuclein. J. Am. Chem. Soc. 2009, 131, 17482–17489.
- Danzer, K.M.; Haasen, D.; Karow, A.R.; Moussaud, S.; Habeck, M.; Giese, A.; Kretzschmar, H.; Hengerer, B.; Kostka, M. Different species of alpha-synuclein oligomers induce calcium influx and seeding. J. Neurosci. 2007, 27, 9220–9232.
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489.
- Volpicelli-Daley, L.A.; Luk, K.C.; Lee, V.M. Addition of exogenous alpha-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous alpha-synuclein to Lewy body and Lewy neurite-like aggregates. Nat. Protoc. 2014, 9, 2135–2146.
- Lashuel, H.A.; Overk, C.R.; Oueslati, A.; Masliah, E. The many faces of α-synuclein: From structure and toxicity to therapeutic target. Nat. Rev. Neurosci. 2013, 14, 38–48.
- Alam, P.; Bousset, L.; Melki, R.; Otzen, D.E. A-Synuclein Oligomers and Fibrils: A Spectrum of Species, a Spectrum of Toxicities. J. Neurochem. 2019, 150, 522–534.
- Volles, M.J.; Lee, S.J.; Rochet, J.C.; Shtilerman, M.D.; Ding, T.T.; Kessler, J.C.; Lansbury, P.T., Jr. Vesicle permeabilization by protofibrillar alpha-synuclein: Implications for the pathogenesis and treatment of Parkinson’s disease. Biochemistry 2001, 40, 7812–7819.
- Pieri, L.; Madiona, K.; Melki, R. Structural and functional properties of prefibrillar alpha-synuclein oligomers. Sci. Rep. 2016, 6, 24526.
- Karpinar, D.P.; Balija, M.B.; Kugler, S.; Opazo, F.; Rezaei-Ghaleh, N.; Wender, N.; Kim, H.Y.; Taschenberger, G.; Falkenburger, B.H.; Heise, H.; et al. Pre-fibrillar alpha-synuclein variants with impaired beta-structure increase neurotoxicity in Parkinson’s disease models. EMBO J. 2009, 28, 3256–3268.
- Winner, B.; Jappelli, R.; Maji, S.K.; Desplats, P.A.; Boyer, L.; Aigner, S.; Hetzer, C.; Loher, T.; Vilar, M.; Campioni, S.; et al. In vivo demonstration that alpha-synuclein oligomers are toxic. Proc. Natl. Acad. Sci. USA 2011, 108, 4194–4199.
- Luth, E.S.; Stavrovskaya, I.G.; Bartels, T.; Kristal, B.S.; Selkoe, D.J. Soluble, prefibrillar α-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J. Biol. Chem. 2014, 289, 21490–21507.
- Ferreira, N.; Gonçalves, N.P.; Jan, A.; Jensen, N.M.; Van Der Laan, A.; Mohseni, S.; Vægter, C.B.; Jensen, P.H. Trans—Synaptic spreading of alpha—Synuclein pathology through sensory afferents leads to sensory nerve degeneration and neuropathic pain. Acta Neuropathol. Commun. 2021, 9, 31.
- Miller, D.W.; Johnson, J.M.; Solano, S.M. Absence of a -synuclein mRNA expression in normal and multiple system atrophy oligodendroglia. J. Neural Transm. 2005, 112, 1613–1624.
- Booth, H.D.E.; Hirst, W.D.; Wade-Martins, R. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 2017, 40, 358–370.
- Lee, H.-J.; Patel, S.; Lee, S.-J. Intravesicular localization and exocytosis of alpha-synuclein and its aggregates. J. Neurosci. 2005, 25, 6016–6024.
- Jang, A.; Lee, H.-J.; Suk, J.-E.; Jung, J.-W.; Kim, K.-P.; Lee, S.-J. Non-classical exocytosis of alpha-synuclein is sensitive to folding states and promoted under stress conditions. J. Neurochem. 2010, 113, 1263–1274.
- Li, J.Y.; Englund, E.; Holton, J.L.; Soulet, D.; Hagell, P.; Lees, A.J.; Lashley, T.; Quinn, N.P.; Rehncrona, S.; Bjorklund, A.; et al. Lewy bodies in grafted neurons in subjects with Parkinson’s disease suggest host-to-graft disease propagation. Nat. Med. 2008, 14, 501–503.
- Kordower, J.H.; Chu, Y.; Hauser, R.A.; Freeman, T.B.; Olanow, C.W. Lewy body-like pathology in long-term embryonic nigral transplants in Parkinson’s disease. Nat. Med. 2008, 14, 504–506.
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113.
- Giguère, N.; Nanni, S.B.; Trudeau, L.E. On cell loss and selective vulnerability of neuronal populations in Parkinson’s disease. Front. Neurol. 2018, 9, 455.
- Engelender, S.; Isacson, O. The Threshold Theory for Parkinson’s Disease. Trends Neurosci. 2017, 40, 4–14.
- Jan, A.; Gonçalves, N.P.; Vaegter, C.B.; Jensen, P.H.; Ferreira, N. The prion-like spreading of alpha-synuclein in parkinson’s disease: Update on models and hypotheses. Int. J. Mol. Sci. 2021, 22, 8338.
- El-Agnaf, O.M.A.; Salem, S.A.; Paleologou, K.E.; Curran, M.D.; Gibson, M.J.; Court, J.A.; Schlossmacher, M.G.; Allsop, D. Detection of oligomeric forms of α-synuclein protein in human plasma as a potential biomarker for Parkinson’s disease. FASEB J. 2006, 20, 419–425.
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35.
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of Disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689.
- Pekny, M.; Pekna, M.; Messing, A.; Steinhäuser, C.; Lee, J.M.; Parpura, V.; Hol, E.M.; Sofroniew, M.V.; Verkhratsky, A. Astrocytes: A central element in neurological diseases. Acta Neuropathol. 2016, 131, 323–345.
- Sofroniew, M. V Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647.
- Tanji, K.; Imaizumi, C.A.T.; Yoshida, H.; Mori, F.; Yoshimoto, M.; Satoh, K.; Wakabayashi, K. Expression of a-synuclein in a human glioma cell line and its up-regulation by interleukin-1 beta. Neuroreport 2001, 12, 1909–1912.
- Chavarría, C.; Rodríguez-bottero, S.; Quijano, C.; Cassina, P.; Souza, J.M. Impact of monomeric, oligomeric and fibrillar alpha-synuclein on astrocyte reactivity and toxicity to neurons. Biochem. J. 2018, 475, 3153–3169.
- Roodveldt, C.; Christodoulou, J.; Dobson, C.M. Immunological features of α-synuclein in Parkinson’s disease. J. Cell. Mol. Med. 2008, 12, 1820–1829.
- Mavroeidi, P.; Xilouri, M. Neurons and glia interplay in α-synucleinopathies. Int. J. Mol. Sci. 2021, 22, 4994.
- Brück, D.; Wenning, G.K.; Stefanova, N.; Fellner, L. Glia and alpha-synuclein in neurodegeneration: A complex interaction. Neurobiol. Dis. 2016, 85, 262–274.
- Lee, H.J.; Kim, C.; Lee, S.J. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxid. Med. Cell. Longev. 2010, 3, 283–287.
- Delaidelli, A.; Richner, M.; Jiang, L.; van der Laan, A.; Bergholdt Jul Christiansen, I.; Ferreira, N.; Nyengaard, J.R.; Vægter, C.B.; Jensen, P.H.; Mackenzie, I.R.; et al. α-Synuclein pathology in Parkinson disease activates homeostatic NRF2 anti-oxidant response. Acta Neuropathol. Commun. 2021, 9, 105.
- Schipper, H.M.; Liberman, A.; Stopa, E.G. Neural heme oxygenase-1 expression in idiopathic Parkinson’s disease. Exp. Neurol. 1998, 150, 60–68.
- Tanji, K.; Maruyama, A.; Odagiri, S.; Mori, F.; Itoh, K.; Kakita, A.; Takahashi, H.; Wakabayashi, K. Keap1 is localized in neuronal and glial cytoplasmic inclusions in various neurodegenerative diseases. J. Neuropathol. Exp. Neurol. 2013, 72, 18–28.
- Ferrer-sueta, G.; Campolo, N.; Trujillo, M.; Bartesaghi, S.; Carballal, S.; Romero, N.; Alvarez, B.; Radi, R. Biochemistry of Peroxynitrite and Protein Tyrosine Nitration. Chem. Rev. 2018, 118, 1338–1408.
- Radi, R. Nitric oxide, oxidants, and protein tyrosine nitration. Proc. Natl. Acad. Sci. USA 2004, 101, 4003–4008.
- Chavarría, C.; Souza, J.M. Oxidation and nitration of alpha-synuclein and their implications in neurodegenerative diseases. Arch. Biochem. Biophys. 2013, 533, 25–32.
- Souza, J.M.; Giasson, B.I.; Chen, Q.; Lee, V.M.Y.; Ischiropoulos, H. Dityrosine cross-linking promotes formation of stable α-synuclein polymers. Implication of nitrative and oxidative stress in the pathogenesis of neurodegenerative synucleinopathies. J. Biol. Chem. 2000, 275, 18344–18349.
- Tsunemi, T.; Ishiguro, Y.; Yoroisaka, A.; Valdez, C.; Miyamoto, K.; Ishikawa, K.; Saiki, S.; Akamatsu, W.; Hattori, N.; Krainc, D. Astrocytes Protect Human Dopaminergic Neurons from a -Synuclein Accumulation and Propagation. J. Neurosci. 2020, 40, 8618–8628.
- Martin, L.J.; Pan, Y.; Price, A.C.; Sterling, W.; Copeland, N.G.; Jenkins, N.A.; Price, D.L.; Lee, M.K. Parkinson’s disease α-synuclein transgenic mice develop neuronal mitochondrial degeneration and cell death. J. Neurosci. 2006, 26, 41–50.
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100.
- Konno, M.; Hasegawa, T.; Baba, T.; Miura, E.; Sugeno, N.; Kikuchi, A.; Fiesel, F.C.; Sasaki, T.; Aoki, M.; Itoyama, Y.; et al. Suppression of dynamin GTPase decreases -synuclein uptake by neuronal and oligodendroglial cells: A potent therapeutic target for synucleinopathy. Mol. Neurodegener. 2012, 7, 38.
- Reyes, J.F.; Rey, N.L.; Bousset, L.; Melki, R.; Brundin, P.; Angot, E. Alpha-synuclein transfers from neurons to oligodendrocytes. Glia 2014, 62, 387–398.
- Lee, Y.; Morrison, B.M.; Li, Y.; Lengacher, S.; Farah, M.H.; Hoffman, P.N.; Liu, Y.; Tsingalia, A.; Jin, L.; Zhang, P.; et al. Oligodendroglia metabolically support axons and contribute to neurodegeneration. Nature 2012, 487, 443–448.
- Funfschilling, U.; Supplie, L.M.; Mahad, D.; Boretius, S.; Aiman, S.; Edgar, J.; Brinkmann, B.G.; Kassmann, C.M.; Tzvetanova, I.D.; Sereda, W.; et al. Glycolytic oligodendrocytes maintain myelin and long-term axonal integrity. Nature 2013, 485, 517–521.
- Philips, T.; Rothstein, J.D. Oligodendroglia: Metabolic supporters of neurons. J. Clin. Investig. 2017, 127, 3271–3280.
- McCann, H.; Stevens, C.H.; Cartwright, H.; Halliday, G.M. α-Synucleinopathy phenotypes. Park. Relat. Disord. 2014, 20, S62–S67.
- Seidel, K.; Mahlke, J.; Siswanto, S.; Krüger, R.; Heinsen, H.; Auburger, G.; Bouzrou, M.; Grinberg, L.T.; Wicht, H.; Korf, H.W.; et al. The brainstem pathologies of Parkinson’s disease and dementia with lewy bodies. Brain Pathol. 2015, 25, 121–135.
- Gilman, S.; Low, P.A.; Quinn, N.; Albanese, A.; Fowler, C.J.; Kaufmann, H.; Klockgether, T.; Lang, A.E.; Lantos, P.L.; Litvan, I.; et al. Consensus statement on the diagnosis of multiple system atrophy. J. Neurol. Sci. 1999, 163, 94–98.
- Mccormack, A.; Chegeni, N.; Chegini, F.; Colella, A.; Power, J.; Keating, D.; Chataway, T. Purification of α-synuclein containing inclusions from human post mortem brain tissue. J. Neurosci. Methods 2016, 266, 141–150.
- Scholz, S.W.; Houlden, H.; Schulte, C.; Sharma, M.; Li, A.; Berg, D.; Melchers, A.; Paudel, R.; Gibbs, J.R.; Simon-Sanchez, J.; et al. SNCA variants are associated with increased risk for multiple system atrophy. Ann. Neurol. 2009, 65, 610–614.
- Kisos, H.; Pukaß, K.; Ben-hur, T.; Richter-landsberg, C.; Sharon, R. Increased Neuronal a -Synuclein Pathology Associates with Its Accumulation in Oligodendrocytes in Mice Modeling a -Synucleinopathies. PLoS ONE 2012, 7, e46817.
- Ubhi, K.; Rockenstein, E.; Mante, M.; Inglis, C.; Adame, A.; Patrick, C.; Whitney, K.; Masliah, E. Neurodegeneration in a Transgenic Mouse Model of Multiple System Atrophy Is Associated with Altered Expression of Oligodendroglial-Derived Neurotrophic Factors. J. Neurosci. 2010, 30, 6236–6246.
- Joers, V.; Tansey, M.; Mulas, G.; Carta, A.R. Microglial phenotypes in Parkinson’s disease and animal models of the disease. Prog. Neurobiol. 2018, 155, 57–75.
- Heneka, M.T.; Golenbock, D.T.; Latz, E. Innate immunity in Alzheimer’s disease. Nat. Immunol. 2015, 16, 229–236.
- Glanzer, J.G.; Enose, Y.; Wang, T.; Kadiu, I.; Gong, N.; Rozek, W.; Liu, J.; Schlautman, J.D.; Ciborowski, P.S.; Thomas, M.P.; et al. Genomic and proteomic microglial profiling: Pathways for neuroprotective inflammatory responses following nerve fragment clearance and activation. J. Neurochem. 2007, 102, 627–645.
- Biber, K.; Neumann, H.; Inoue, K.; Boddeke, H.W.G.M. Neuronal ‘ On ’ and ‘ Off ’ signals control microglia. Trends Neurosci. 2007, 30, 596–602.
- Mackenzie, I.R.A. Activated microglia in dementia with Lewy bodies. Neurology 2000, 55, 132–135.
- Ferreira, S.A.; Romero-ramos, M. Microglia Response During Parkinson’s Disease: Alpha-Synuclein Intervention. Front. Cell Neurosci. 2018, 12, 247.
- Theodore, S.; Shuwen Cao, B.; McLean, P.J.; Standaert, D. Targeted Overexpression of Human Alpha-Synuclein Triggers Microglial Activation and an Adaptive Immune Response in a Mouse Model of Parkinson Disease. J. Neuropathol. Exp. Neurol. 2009, 67, 1149–1158.
- Béraud, D.; Twomey, M.; Bloom, B.; Mittereder, A.; Ton, V.; Neitzke, K.; Chasovskikh, S.; Mhyre, T.R.; Maguire-Zeiss, K.A. α-Synuclein Alters Toll-Like Receptor Expression. Front. Neurosci. 2011, 5, 80.
- Hou, L.; Bao, X.; Zang, C.; Yang, H.; Sun, F.; Che, Y.; Wu, X.; Li, S.; Zhang, D.; Wang, Q. Integrin CD11b mediates α-synuclein-induced activation of NADPH oxidase through a Rho-dependent pathway. Redox Biol. 2018, 14, 600–608.
- Stefanova, N.; Reindl, M.; Neumann, M.; Kahle, P.J.; Poewe, W.; Wenning, G.K. Microglial Activation Mediates Neurodegeneration Related to Oligodendroglial alpha -Synucleinopathy: Implications for Multiple System Atrophy. Mov. Disord. 2007, 22, 2196–2203.
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2021, 35, 441–468.
- Park, J.; Paik, S.R.; Jou, I.L.O.; Park, S.M. Microglial Phagocytosis Is Enhanced by Monomeric a -Synuclein, Not Aggregated a -Synuclein: Implications for Parkinson’s Disease. Glia 2008, 1223, 1215–1223.
- Zhang, W.; Wang, T.; Pei, Z.; Miller, D.S.; Wu, X.; Block, M.L.; Wilson, B.; Zhang, W.; Zhou, Y.; Hong, J.-S.; et al. Aggregated -synuclein activates microglia: A process leading to disease progression in Parkinson’s disease. FASEB J. 2005, 19, 533–542.
- Sarkar, S.; Dammer, E.B.; Malovic, E.; Olsen, A.L.; Raza, S.A.; Gao, T.; Xiao, H.; Oliver, D.L.; Duong, D.; Joers, V.; et al. Molecular Signatures of Neuroinflammation Induced by α Synuclein Aggregates in Microglial. Front. Immunol. 2020, 11, 33.
- Galatro, T.F.; Holtman, I.R.; Lerario, A.M.; Vainchtein, I.D.; Brouwer, N.; Sola, P.R.; Veras, M.M.; Pereira, T.F.; Leite, R.E.P.; Möller, T.; et al. Transcriptomic analysis of purified human cortical microglia reveals age-associated changes. Nat. Neurosci. 2017, 20, 1162–1171.
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; Keeffe, S.O.; Phatnani, H.P.; Guarnieri, X.P.; Caneda, C.; Ruderisch, N.; et al. An RNA-Sequencing Transcriptome and Splicing Database of Glia, Neurons, and Vascular Cells of the Cerebral Cortex. J. Neurosci. 2014, 34, 11929–11947.
- Angelopoulou, E.; Paudel, Y.N.; Villa, C.; Shaikh, M.F.; Piperi, C. Lymphocyte-activation gene 3 (LAG3) protein as a possible therapeutic target for Parkinson’s disease: Molecular mechanisms connecting neuroinflammation to α-synuclein spreading pathology. Biology 2020, 9, 86.
- Mao, X.; Ou, M.T.; Karuppagounder, S.S.; Kam, T.-I.; Yin, X.; Xiong, Y.; Ge, P.; Essien Umanah, G.; Brahmachari, S.; Shin, J.-H.; et al. Pathological α-synuclein transmission initiated by binding lymphocyte-activation gene 3. Science 2016, 353, aah3374.
- Aulić, S.; Masperone, L.; Narkiewicz, J.; Isopi, E.; Bistaffa, E.; Pastore, B.; De Cecco, E.; Scaini, D.; Zago, P.; Moda, F.; et al. α-Synuclein Amyloids Hijack Prion Protein to Gain Cell Entry, Facilitate Cell-to-Cell Spreading and Block Prion Replication. Sci. Rep. 2017, 7, 10050.
- Ferreira, D.G.; Temido-Ferreira, M.; Miranda, H.V.; Batalha, V.L.; Coelho, J.E.; Szegö, É.M.; Marques-Morgado, I.; Vaz, S.H.; Rhee, J.S.; Schmitz, M.; et al. α-Synuclein interacts with PrP C to induce cognitive impairment through mGluR5 and NMDAR2B. Nat. Neurosci. 2017, 20, 1569–1579.
- Ihse, E.; Yamakado, H.; Van Wijk, X.M.; Lawrence, R.; Esko, J.D. Cellular internalization of alpha- synuclein aggregates by cell surface heparan sulfate depends on aggregate conformation and cell type. Sci. Rep. 2017, 7, 9008.
- Stefanova, N.; Fellner, L.; Reindl, M.; Masliah, E.; Poewe, W.; Wenning, G.K. Toll-like receptor 4 promotes α-synuclein clearance and survival of nigral dopaminergic neurons. Am. J. Pathol. 2011, 179, 954–963.
- Kim, C.; Ho, D.; Suk, J.; You, S.; Michael, S.; Kang, J.; Lee, S.J.; Masliah, E.; Hwang, D.; Lee, H.; et al. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nat. Commun. 2014, 4, 1562.
- Shrivastava, A.N.; Redeker, V.; Fritz, N.; Pieri, L.; Almeida, L.G.; Spolidoro, M.; Liebmann, T.; Bousset, L.; Renner, M.; Léna, C.; et al. a-synuclein assemblies sequester neuronal a3-Na+/K+-ATPase and impair Na+ gradient Amulya. EMBO J. 2015, 34, 2408–2423.
- Choi, Y.R.; Cha, S.H.; Kang, S.J.; Kim, J.B.; Jou, I.; Park, S.M. Prion-like Propagation of α-Synuclein Is Regulated by the FcγRIIB-SHP-1/2 Signaling Pathway in Neurons. Cell Rep. 2018, 22, 136–148.