Propofol or barbiturate application at low concentrations increases desensitization and slows deactivation of GABA-induced current and propofol/barbiturate at high concentrations directly elicits after-responses upon their washout in hippocampal or sensory neurons. It is postulated that the generation of such after-responses is caused by removal of the blockade by anesthetic agents as partial antagonists. However, the increased desensitization was invariably followed by slowdown of deactivation of GABA-induced current, and the after-response may arise as a consequence of extreme slowdown of deactivation following strong desensitization. It is thus possible that propofol and barbiturate can facilitate resensitization of GABA responses. Propofol and barbiturate are useful to treat the alcohol/benzodiazepine withdrawal syndrome. Considering that the slowdown of deactivation following desensitization and the after-response induced by propofol or barbiturate application, the regulatory mechanisms of desensitization/resensitization of GABAAR-mediated currents might be important for understanding the treatment of the alcohol/benzodiazepine withdrawal syndrome.
- GABA
- Desensitization
- Resensitization
Alcohol/benzodiazepine withdrawal syndrome and its treatment by modulating desensitization/resensitization kinetics of GABAAR-mediated currents
Alcohol and benzodiazepine are useful to mitigate anxiety through enhancing GABAAR-mediated inhibition. However, alcohol and benzodiazepine are known as abused drugs. The alcohol or benzodiazepine withdrawal syndrome appears following a reduction on alcohol or benzodiazepine use after a period of excessive use [1][2][3][4]. The alcohol or benzodiazepine withdrawal symptoms typically include anxiety, sweating, hand tremor and sleep disturbance. The underlying mechanisms involve neuronal adaptations, which are revealed as decreased GABAergic responses [5] and enhancement of NMDA responses [6][7][8][9]. Although the exact mechanism for the reduced responsiveness of GABAARs remains uncertain, changes in surface GABAAR protein level and subunit composition, changes in turnover, recycling, and production rates, degree of phosphorylation and decreased coupling mechanisms between GABA and alcohol/benzodiazepine sites are thought to be involved in the reduced responsiveness [10][11][12][13]. It has recently been demonstrated that the benzodiazepine diazepam caused downregulation of GABAergic inhibition through phospholipase C (PLCδ)/Ca2+/calcineurin signaling pathway [14]. The study showed that overexpression of PRIP-1 suppressed diazepam-dependent activation of PLCδ and diazepam-dependent downregulation of GABAARs in HEK293 cells [14], indicating that PRIP-1 acts as an inhibitor by outcompeting the PLCδ binding to GABAARs. Because intracellular Ca2+ and calcineurin activity are increased in PRIP-DKO mice [15], these findings suggest that the diazepam-induced long-term downregulation of GABAergic inhibition is mediated by PLCδ/Ca2+/calcineurin signaling pathway. Nevertheless, it is also true that calcineurin causes resensitization of GABAAR-mediated currents by facilitating their desensitization [15][16]. Given the apparently contradictory behaviors of GABAAR-mediated currents by calcineurin activation, the two different behaviors of GABAAR-mediated currents may depend on whether calcineurin activation occurs before or after activation of GABAARs.
As for the treatment of the alcohol/benzodiazepine withdrawal syndrome, propofol and barbiturate which enhance GABAAR-mediated inhibition are useful. Indeed, it was demonstrated that propofol and barbiturates (pentobarbital and phenobarbital) were effective for the treatment of alcohol withdrawal syndrome [17][18], and barbiturate (pentobarbital) was effective for the treatment of benzodiazepine withdrawal syndrome [19]. However, it remains unclear how propofol and barbiturate ameliorate reduced GABA responsiveness in patients with alcohol/benzodiazepine withdrawal syndrome. Although the concentrations of propofol and barbiturates that generated the hump-like current are very high [20][21][22] compared to the dose used for treatment of the withdrawal syndrome [17][18], the generation of hump-like GABAAR currents itself may suggest the occurrence of resensitization of GABAAR-mediated currents. Indeed, the desensitization and deactivation of GABAAR-mediated currents are facilitated and slowed, respectively, by propofol/barbiturate at much lower concentrations [21][22]. Then, propofol and barbiturate may improve the reduced GABA responsiveness through the resensitization of GABAAR-mediated currents. Therefore, the regulatory mechanisms of desensitization/resensitization of GABAAR-mediated currents are important to better understand the benzodiazepine/alcohol withdrawal syndrome and to develop the treatment method.
Drugs that cause desensitization and resensitization of GABAAR-mediated currents
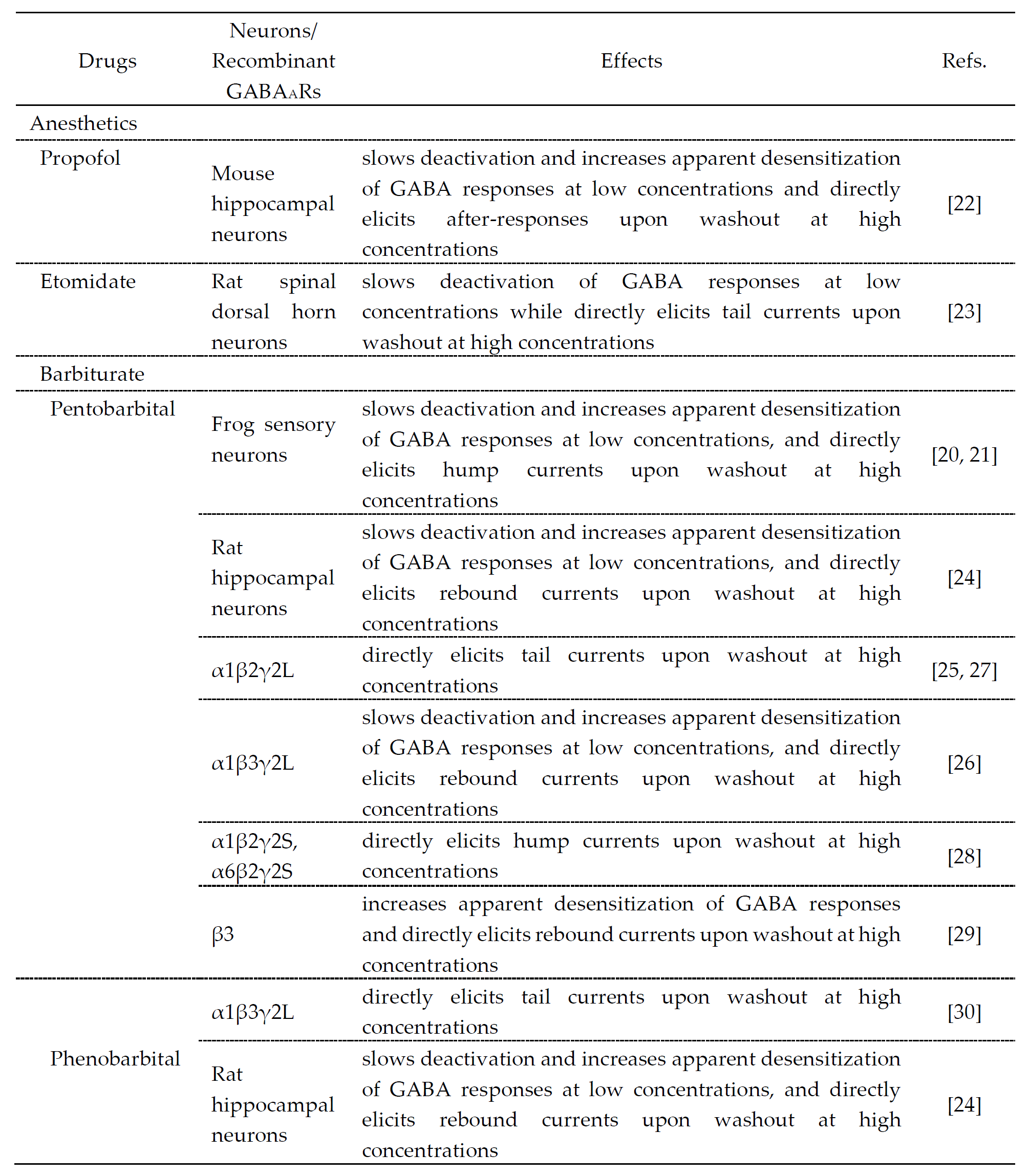
The drugs that cause desensitization and resensitization of GABAAR-mediated currents are summarized in Table 1. Hump-like GABAAR currents after a strong desensitization were seen at the offset of propofol application at a high concentration (600 μM) in hippocampal pyramidal neurons [22], etomidate application at a high concentration (1 mM) in rat spinal dorsal horn neurons [23], pentobarbital application at high concentrations (1–3 mM) in frog sensory neurons [21][22], rat hippocampal neurons [24] and recombinant GABAARs [25][26][27][28][29][30] or phenobarbital application at a high concentration (10 mM) in rat hippocampal neurons [24], although these were not seen at the offset of GABA application. It is believed that the generation of hump-like currents may be caused by removal of the blockade by anesthetic agents as partial antagonists [25], although their mechanisms remain unclear and the involvement of desensitization is not necessarily denied. To better understand the actions of these anesthetic drugs, the molecular and regulatory mechanisms of desensitization/resensitization of GABAAR-mediated currents might be important.
Table 1. Drugs that modulate GABA responses and directly activate GABAARs at higher concentrations.
A possible kinetic mechanism for resensitization of GABAA currents
In layer II/III pyramidal cells of the mouse barrel cortex, the deletion of PRIP-1/2 enhanced the desensitization of GABAAR-mediated currents but paradoxically induced a hump-like tail-current at the offset of the GABA puff [15], and also resulted in the prolongation of the decay phase of eIPSCs [31]. To understand the kinetic mechanisms underlying the generation of the hump-like tail-currents and the prolongation of eIPSCs, these currents were simulated using a model previously proposed [32] (Figure 1). It was examined whether the possible increase in the fast desensitization rate (d2) and the possible decrease in the unbinding rate (koff) can lead to the generation of the hump-like tail-current at the offset of the GABA puff. In the simulated wild-type pyramidal cell, GABAAR-mediated currents were induced without a hump-like tail-current in response to 2-sec GABA puff at 50 µM [15]. In contrast, in the simulated PRIP-DKO pyramidal cell, GABAAR-mediated currents displayed a prominent desensitization and was followed by a prominent hump-like tail-current [15]. Thus, a slowdown of koff and an acceleration of d2 resulted in a generation of a hump-like tail-current. Following a sharp decrease in [GABA] at the offset of GABA puff, a sharp decrease in d2 to a level smaller than the fast de-desensitization (i.e. resensitization) rate constant (r2) occurred to subsequently induce a hump-like tail-current. It can be concluded that a higher calcineurin activity in PRIP-DKO layer III pyramidal cells might have caused a slowdown of koff and an acceleration of d2 through the modulation of its GABA concentration dependency, leading to a generation of hump-like tail-currents in PRIP-DKO pyramidal cells. It was also investigated whether the increase in d2 and the decrease in koff can also lead to the prolongation of eIPSCs. Simulated IPSCs in PRIP-DKO and the wild-type pyramidal cells that have the half-durations similar to those obtained in the real experiments [15] revealed that a prolongation of eIPSCs/eIPSPs in PRIP-DKO pyramidal cells results from resensitization of GABAAR-mediated currents, which is brought about by an acceleration of d2 through the modulation of its [GABA] dependency together with a slowdown of koff.
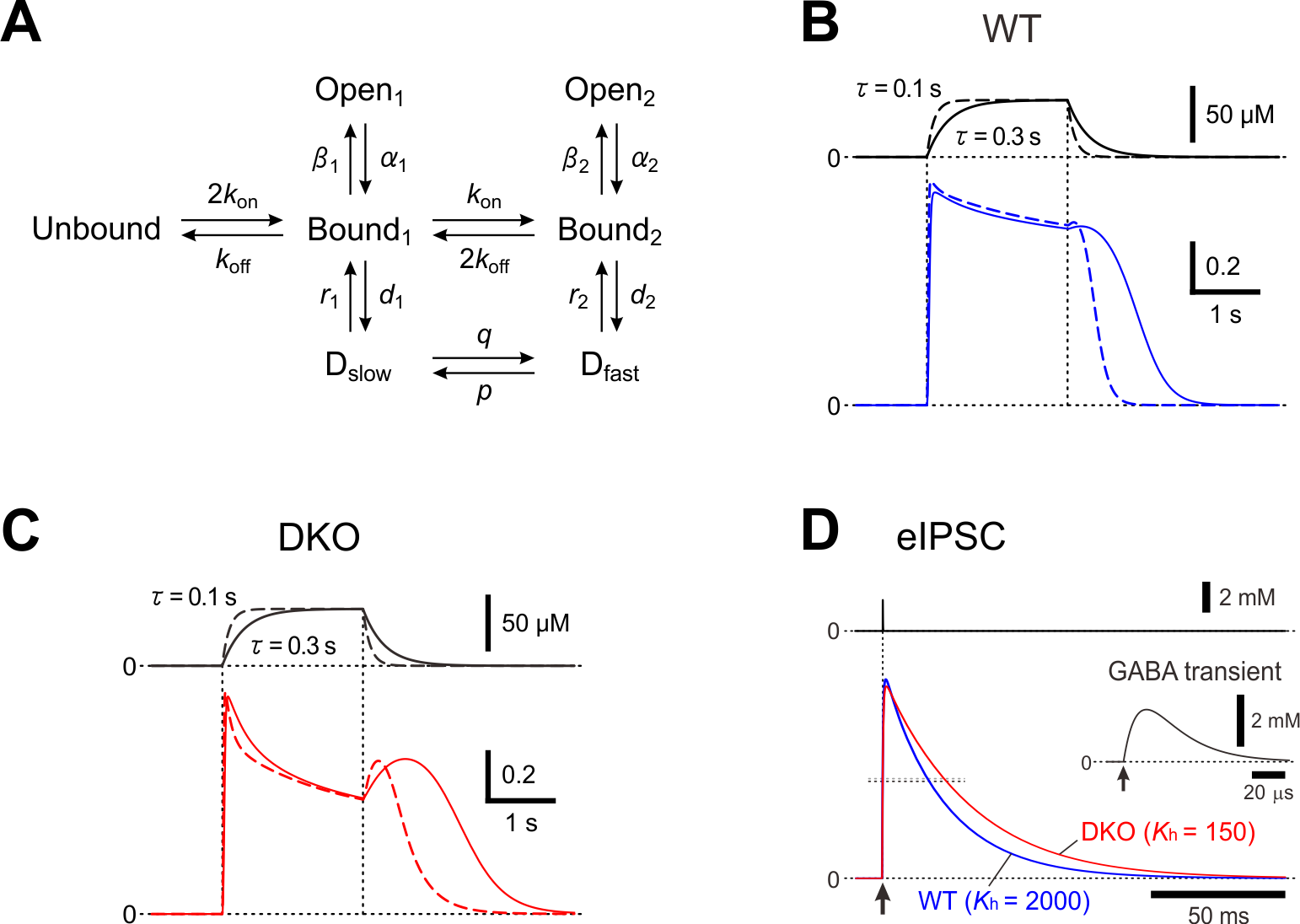
Figure 1. A kinetic model for a hump-like tail-current. (A) A kinetic model of GABAARs representing mono- and double-liganded states, each providing access to open and desensitized states. (B and C) Top; Presumed [GABA] changes created by puff application of GABA with a rectangular pressure pulse through a puff pipette containing 200 μM GABA in the extracellular medium was assumed to be diluted 4 times, and the onset and offset of the puff application were assumed to be attenuated with a time constant raging between 0.1 and 0.3 sec. Bottom; superimposed traces of the simulated GABAAR-mediated currents under the condition that the attenuation time constant is 0.3 and 0.1 sec (solid and interrupted traces, respectively) in simulated wild-type (B) and PRIP-DKO (C) pyramidal cells. (D) Superimposed traces of a simulated wild-type and PRIP-DKO eIPSC induced by a GABA transient shown on an expanded time scale (inset) with a small maximum conductance. Adopted from [15] and [31].
Based on the experimental and simulation studies, the regulatory mechanisms of GABAARs are schematically depicted (Figure 2).
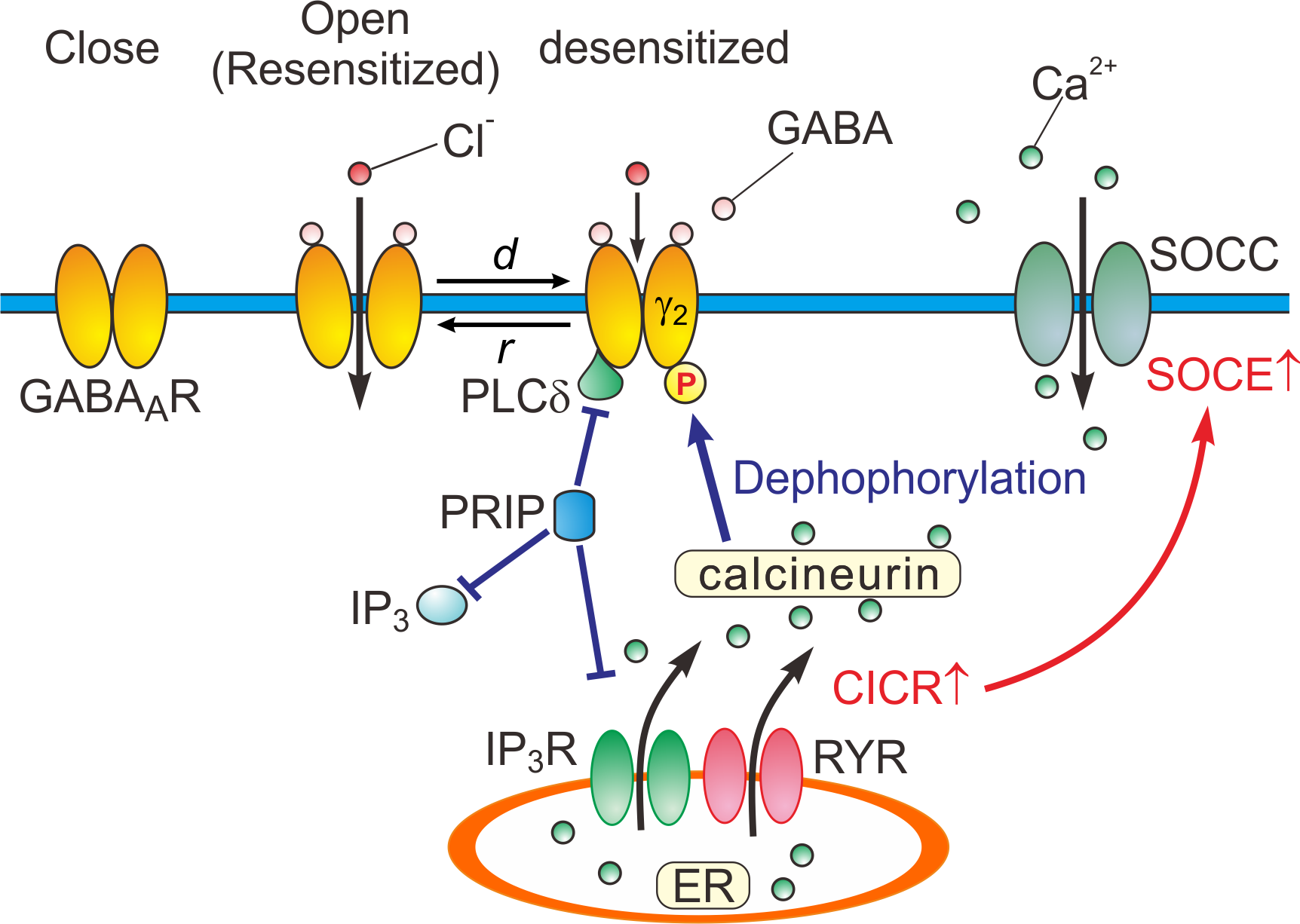
Figure 2. Close, open (resensitized) and desensitized states of GABAARs. When GABA binds to GABAARs, the receptors open the pore, and consequently increasing the permeability of the ion pore to Cl-. In response to a prolonged application of GABA, GABAARs are desensitized (d) by increased calcineurin activity due to potentiated Ca2+-induced Ca2+ release (CICR) followed by store-operated Ca2+ entry (SOCE) [15]. GABAARs are resensitized through the de-desensitization (r) at the offset of the GABA puff. PRIP outcompetes the PLCδ in binding to GABAAR β subunits [14]. d: desensitization, r: resensitization, RYR: ryanodine receptor, SOCC: store-operated Ca2+ channel, IP3R: inositol trisphosphate receptor.
References
- Bayard, M.; McIntyre, J.; Hill, K. R.; Woodside, J., Jr.; Alcohol withdrawal syndrome. Am Fam Physician 2004, 69, (6), 1443-50, .
- A McKeon; M A Frye; Norman Delanty; The alcohol withdrawal syndrome. Journal of Neurology, Neurosurgery & Psychiatry 2008, 79, 854-862, 10.1136/jnnp.2007.128322.
- S R Onyett; The benzodiazepine withdrawal syndrome and its management.. The Journal of the Royal College of General Practitioners 1989, 39, 160-163, .
- H. Pétursson; The benzodiazepine withdrawal syndrome. Addiction 1994, 89, 1455-1459, 10.1111/j.1360-0443.1994.tb03743.x.
- C Allison; J A Pratt; Neuroadaptive processes in GABAergic and glutamatergic systems in benzodiazepine dependence.. Pharmacology & Therapeutics 2003, 98, 171-195, 10.1016/s0163-7258(03)00029-9.
- L. Judson Chandler; Hunter Newsom; Colin Sumners; Fulton Crews; Chronic Ethanol Exposure Potentiates NMDA Excitotoxicity in Cerebral Cortical Neurons. Journal of Neurochemistry 1993, 60, 1578-1581, 10.1111/j.1471-4159.1993.tb03326.x.
- Kathleen A. Grant; Peter Valverius; Michael Hudspith; Boris Tabakoff; Ethanol withdrawal seizures and the NMDA receptor complex. European Journal of Pharmacology 1990, 176, 289-296, 10.1016/0014-2999(90)90022-x.
- Jonathan M. Koff; Gary A. Pritchard; David J. Greenblatt; Lawrence G. Miller; The NMDA receptor competitive antagonist CPP modulates benzodiazepine tolerance and discontinuation.. Pharmacology 1997, 55, 217-227, 10.1159/000139531.
- Makoto Tsuda; Norifumi Shimizu; Yoshinori Yajima; T. Suzuki; Miwa Misawa; Hypersusceptibility to DMCM-induced seizures during diazepam withdrawal in mice: evidence for upregulation of NMDA receptors.. Naunyn-Schmiedeberg's Archives of Pharmacology 1998, 357, 309-315, 10.1007/pl00005172.
- Richard W. Olsen; Jing Liang; Elisabetta Cagetti; Igor Spigelman; Plasticity of GABAA Receptors in Brains of Rats Treated with Chronic Intermittent Ethanol. Neurochemical Research 2005, 30, 1579-1588, 10.1007/s11064-005-8836-6.
- Richard W. Olsen; Igor Spigelman; GABAA receptor plasticity in alcohol withdrawal. Epilepsia 2010, 51, 50-50, 10.1111/j.1528-1167.2010.02836.x.
- Kelly R. Tan; Uwe Rudolph; Christian Lüscher; Hooked on benzodiazepines: GABAA receptor subtypes and addiction.. Trends in Neurosciences 2011, 34, 188-97, 10.1016/j.tins.2011.01.004.
- Keith A Wafford; GABAA receptor subtypes: any clues to the mechanism of benzodiazepine dependence?. Current Opinion in Pharmacology 2005, 5, 47-52, 10.1016/j.coph.2004.08.006.
- Martin W. Nicholson; Aaron Sweeney; Eva Pekle; Sabina Alam; Afia B. Ali; Michael Duchen; Jasmina N. Jovanovic; Diazepam-induced loss of inhibitory synapses mediated by PLCδ/ Ca2+/calcineurin signalling downstream of GABAA receptors.. Molecular Psychiatry 2018, 23, 1851-1867, 10.1038/s41380-018-0100-y.
- Hiroki Toyoda; Mitsuru Saito; Hajime Sato; Takuma Tanaka; Takeo Ogawa; Hirofumi Yatani; Tsutomu Kawano; Takashi Kanematsu; Masato Hirata; Youngnam Kang; et al. Enhanced desensitization followed by unusual resensitization in GABAA receptors in phospholipase C-related catalytically inactive protein-1/2 double-knockout mice. Pflügers Archiv - European Journal of Physiology 2014, 467, 267-284, 10.1007/s00424-014-1511-5.
- Mathew V. Jones; Gary L. Westbrook; Shaping of IPSCs by Endogenous Calcineurin Activity. The Journal of Neuroscience 1997, 17, 7626-7633, 10.1523/JNEUROSCI.17-20-07626.1997.
- Amy L. Brotherton; Eric P. Hamilton; H. Grace Kloss; Drayton A. Hammond; Propofol for Treatment of Refractory Alcohol Withdrawal Syndrome: A Review of the Literature. Pharmacotherapy: The Journal of Human Pharmacology and Drug Therapy 2016, 36, 433-442, 10.1002/phar.1726.
- Katherine Martin; Andrew Katz; The Role of Barbiturates for Alcohol Withdrawal Syndrome. Journal of Psychosomatic Research 2016, 57, 341-347, 10.1016/j.psym.2016.02.011.
- S. H. Preskorn; Benzodiazepines and withdrawal psychosis. Report of three cases. JAMA 1977, 237, 36-38, 10.1001/jama.237.1.36.
- N Akaike; K Hattori; N Inomata; Y Oomura; gamma-Aminobutyric-acid- and pentobarbitone-gated chloride currents in internally perfused frog sensory neurones.. The Journal of Physiology 1985, 360, 367-386, 10.1113/jphysiol.1985.sp015622.
- N Akaike; T Maruyama; N Tokutomi; Kinetic properties of the pentobarbitone-gated chloride current in frog sensory neurones.. The Journal of Physiology 1987, 394, 85-98, 10.1113/jphysiol.1987.sp016861.
- Ba Orser; Ly Wang; Ps Pennefather; Jf Macdonald; Propofol modulates activation and desensitization of GABAA receptors in cultured murine hippocampal neurons. The Journal of Neuroscience 1994, 14, 7747-7760, 10.1523/JNEUROSCI.14-12-07747.1994.
- Zhen-Xiong Zhang; Hui Lü; Xian-Ping Dong; Jin Liu; Tian-Le Xu; Kinetics of etomidate actions on GABA(A) receptors in the rat spinal dorsal horn neurons.. Brain Research 2002, 953, 93-100, .
- J M Rho; S D Donevan; M A Rogawski; Direct activation of GABAA receptors by barbiturates in cultured rat hippocampal neurons.. The Journal of Physiology 1996, 497, 509-522, 10.1113/jphysiol.1996.sp021784.
- Kevin J. Gingrich; Paul M. Burkat; William A. Roberts; Pentobarbital Produces Activation and Block of α1β2γ2S GABAA Receptors in Rapidly Perfused Whole Cells and Membrane Patches: Divergent Results Can Be Explained by Pharmacokinetics. The Journal of General Physiology 2009, 133, 171-188, 10.1085/jgp.200810081.
- Hua-Jun Feng; Matt T. Bianchi; Robert L. Macdonald; Pentobarbital Differentially Modulates α1β3δ and α1β3γ2L GABAA Receptor Currents. Molecular Pharmacology 2004, 66, 988-1003, 10.1124/mol.104.002543.
- Klaus Krampfl; Heiner Wolfes; Reinhard Dengler; Johannes Bufler; Kinetic analysis of the agonistic and blocking properties of pentobarbital on recombinant rat alpha(1)beta(2)gamma(2S) GABA(A) receptor channels.. European Journal of Pharmacology 2002, 435, 1-8, .
- S.A. Thompson; P.J. Whiting; K.A. Wafford; Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. British Journal of Pharmacology 1996, 117, 521-527, 10.1111/j.1476-5381.1996.tb15221.x.
- Julian R. A. Wooltorton; Stephen J. Moss; Trevor G. Smart; Pharmacological and Physiological Characterization of Murine Homomeric β3 GABAAReceptors. European Journal of Neuroscience 1997, 9, 2225-2235, 10.1111/j.1460-9568.1997.tb01641.x.
- Alexis M. Ziemba; Stuart A. Forman; Correction for Inhibition Leads to an Allosteric Co-Agonist Model for Pentobarbital Modulation and Activation of α1β3γ2L GABAA Receptors. PLOS ONE 2016, 11, e0154031, 10.1371/journal.pone.0154031.
- Hiroki Toyoda; Mitsuru Saito; Hajime Sato; Tsutomu Kawano; Shinpei Kawakami; Hirofumi Yatani; Takashi Kanematsu; Masato Hirata; Youngnam Kang; Enhanced lateral inhibition in the barrel cortex by deletion of phospholipase C-related catalytically inactive protein-1/2 in mice. Pflügers Archiv - European Journal of Physiology 2014, 467, 1445-1456, 10.1007/s00424-014-1592-1.
- Mathew V. Jones; Gary L. Westbrook; Desensitized states prolong GABAA channel responses to brief agonist pulses. Neuron 1995, 15, 181-191, 10.1016/0896-6273(95)90075-6.